
Risk Factors Associated with the Clinical Outcomes of COVID-19 and Its Variants in the Context of Cytokine Storm and Therapeutics/Vaccine Development Challenges

 and
and
Abstract
:1. Background
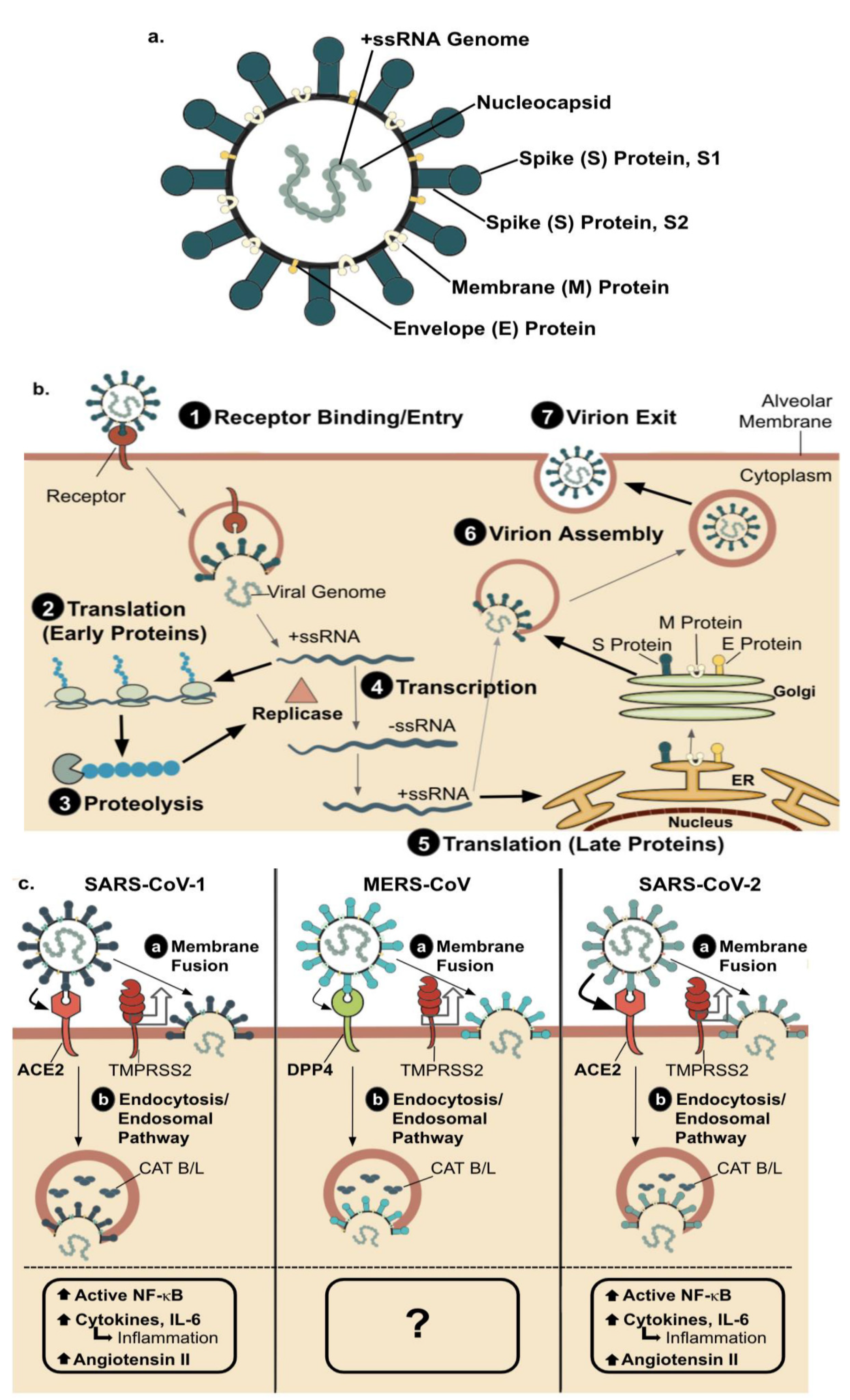
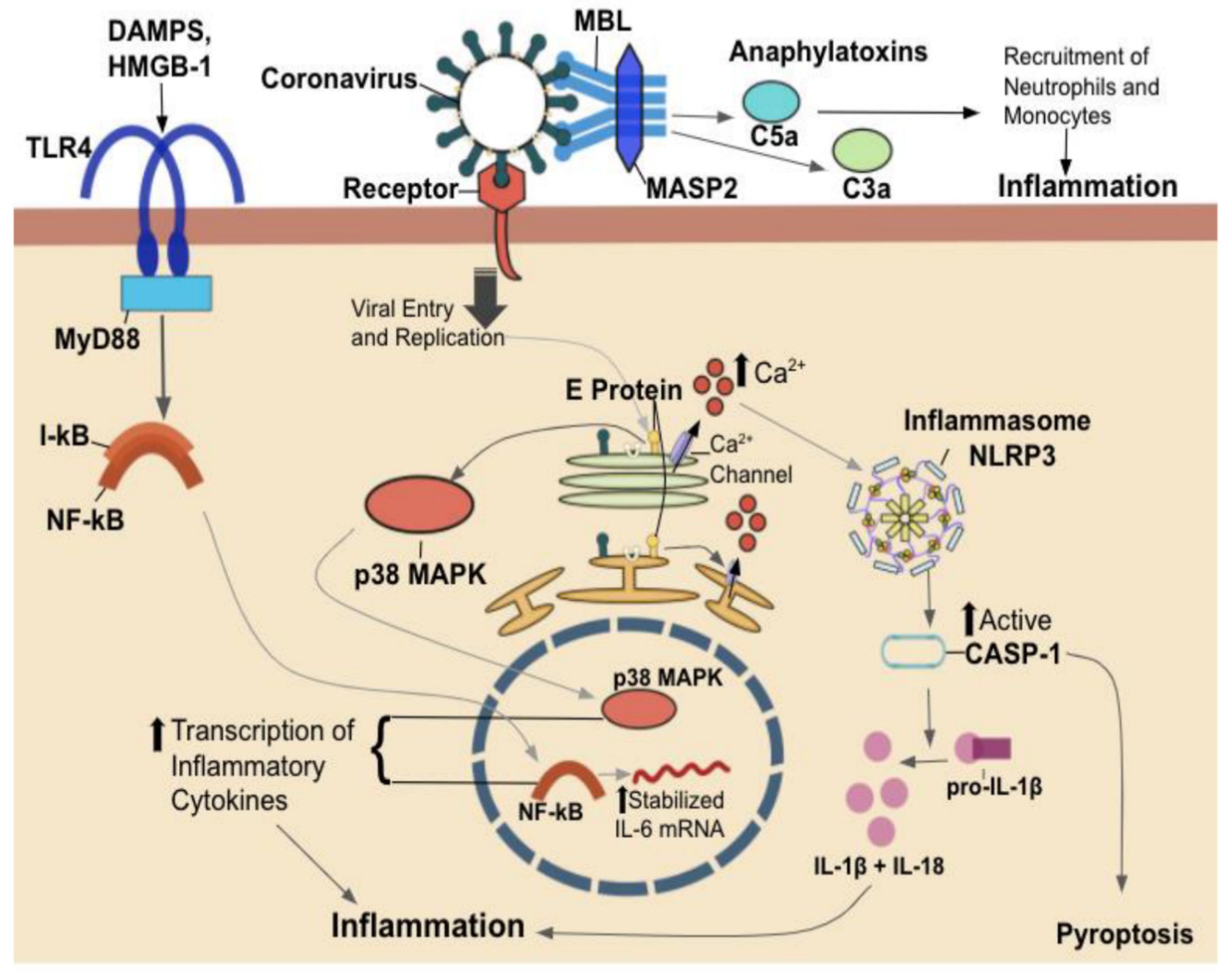
2. Host-Pathogen Interaction
3. Risk Factors
3.1. Sex and Race
3.2. Genetic Factors
3.2.1. HLA Polymorphisms and Disease Manifestations
3.2.2. HLA-DR Expression and Disease Manifestations
3.2.3. Blood Type
3.3. Pre-Existing Conditions and Vulnerabilities
3.3.1. Vitamin D Deficiency
3.3.2. Obesity
3.3.3. Smoking, Nicotine, and Asthma
4. Therapies and Vaccines
Vaccines
5. Conclusions
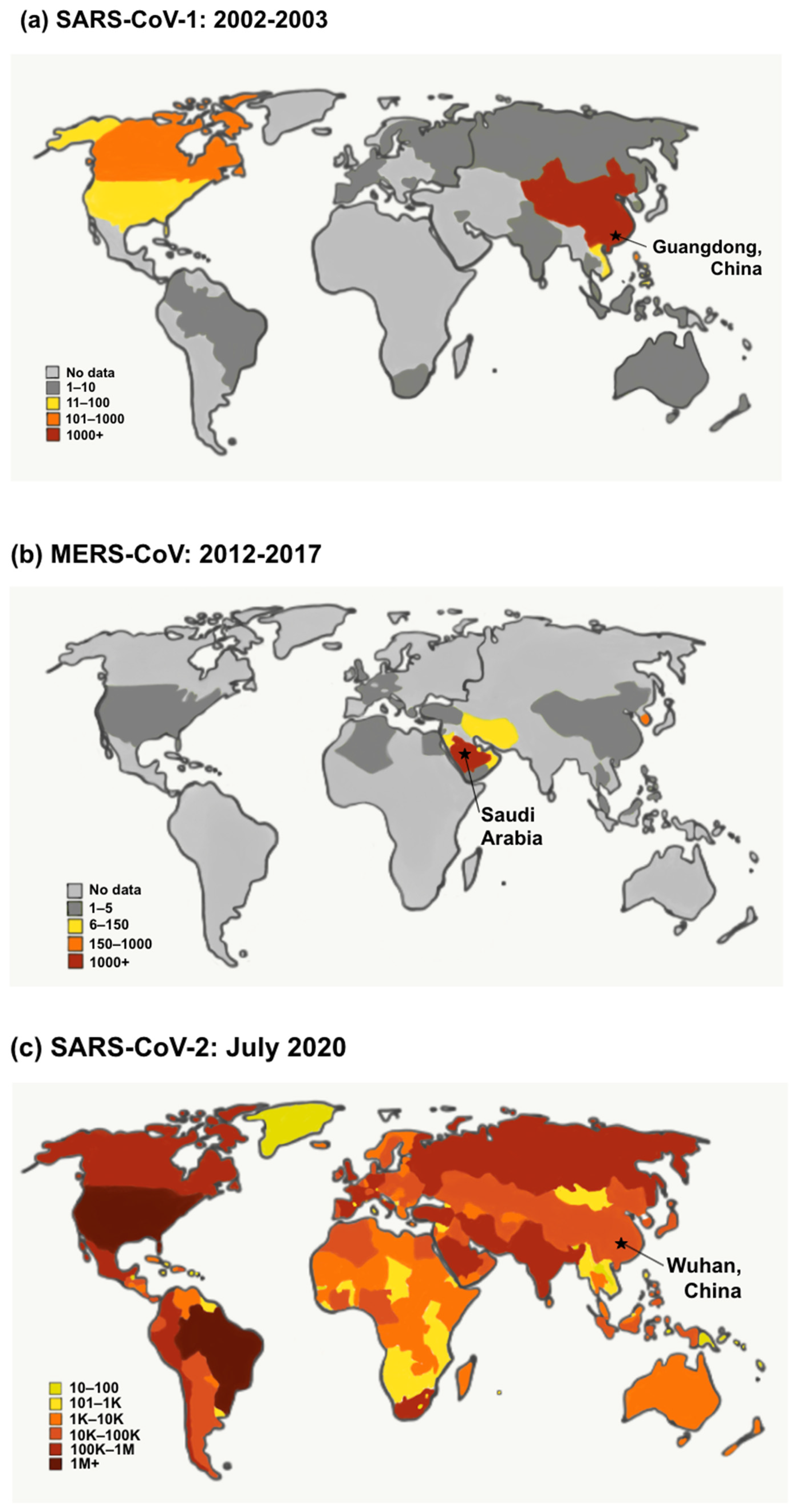
6. Gaining Perspective: Lessons from the Existing Coronaviruses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| Coronavirus disease 2019 | COVID-19 |
| Dendritic cells | DCs |
| Human leukocyte antigen | HLA |
| Severe acute respiratory syndrome | SARS |
| Middle East respiratory syndrome | MERS |
| Receptor-binding domain | RBD |
| Positive-sense/negative-sense single-stranded RNA | +/-ssRNA |
| Interferon | IFN |
| Angiotensin-converting enzyme 2 | ACE2 |
| Dipeptidyl peptidase 4 | DPP4 |
| Acute respiratory distress syndrome | ARDS |
| Pathogen-associated molecular patterns | PAMPS |
| Pattern recognition receptors | PRRs |
| Toll-like receptors | TLRs |
| Dendritic cell-specific ICAM-3-grabbing non-integrin | DC-SIGN |
| Natural killer cell | NK Cell |
| Myeloid differentiation primary response 88 | MyD88 |
| Nuclear factor-kappa B | NF-κB |
| Angiotensin II | Ang II |
| C-reactive protein | CRP |
| Tuberculosis | TB |
| Body mass index | BMI |
| Lopinavir | LPV |
| Ritonavir | RTV |
| Chlorpromazine | CPZ |
| Modified vaccinia virus Ankara | MVA |
| Vitamin A deficiency | VAD |
| COVID-19-associated mucormycosis | CAM |
References
- Fani, M.; Teimoori, A.; Ghafari, S. Comparison of the COVID-2019 (SARS-CoV-2) pathogenesis with SARS-CoV and MERS-CoV infections. Future Virol. 2020, 15, 317–323. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Li, X.; Li, T.; Zhang, S.; Wang, L.; Wu, X.; Liu, J. The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 1–7. [Google Scholar] [CrossRef]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- Abduljalil, J.M.; Abduljalil, B.M. Epidemiology, genome, and clinical features of the pandemic SARS-CoV-2: A recent view. New Microbes New Infect. 2020, 35, 100672. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Ramirez, S.I.; Lokugamage, K.G.; Makino, S. Coronavirus nonstructural protein 1: Common and distinct functions in the regulation of host and viral gene expression. Virus Res. 2015, 202, 89–100. [Google Scholar] [CrossRef]
- Neogi, U.; Hill, K.J.; Ambikan, A.T.; Heng, X.; Quinn, T.P.; Byrareddy, S.N.; Sonnerborg, A.; Sarafianos, S.G.; Singh, K. Feasibility of Known RNA Polymerase Inhibitors as Anti-SARS-CoV-2 Drugs. Pathogens 2020, 9, 320. [Google Scholar] [CrossRef]
- Hui, D.S. Epidemic and Emerging Coronaviruses (Severe Acute Respiratory Syndrome and Middle East Respiratory Syndrome). Clin. Chest Med. 2017, 38, 71–86. [Google Scholar] [CrossRef]
- Meyerholz, D.K.; Lambertz, A.M.; McCray, P.B. Dipeptidyl Peptidase 4 Distribution in the Human Respiratory Tract: Implications for the Middle East Respiratory Syndrome. Am. J. Pathol. 2016, 186, 78–86. [Google Scholar] [CrossRef] [Green Version]
- van den Brand, J.M.; Smits, S.L.; Haagmans, B.L. Pathogenesis of Middle East respiratory syndrome coronavirus. J. Pathol. 2015, 235, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Dyall, J.; Gross, R.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.G., Jr.; Hensley, L.E.; Frieman, M.B.; Jahrling, P.B. Middle East Respiratory Syndrome and Severe Acute Respiratory Syndrome: Current Therapeutic Options and Potential Targets for Novel Therapies. Drugs 2017, 77, 1935–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubernatorova, E.O.; Gorshkova, E.A.; Polinova, A.I.; Drutskaya, M.S. IL-6: Relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev. 2020, 53, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geijtenbeek, T.B.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Adema, G.J.; van Kooyk, Y.; Figdor, C.G. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell 2000, 100, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.; Thomsen, A.R. Sensing of RNA Viruses: A Review of Innate Immune Receptors Involved in Recognizing RNA Virus Invasion. J. Virol. 2012, 86, 2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- Moreno-Eutimio, M.A.; López-Macías, C.; Pastelin-Palacios, R. Bioinformatic analysis and identification of single-stranded RNA sequences recognized by TLR7/8 in the SARS-CoV-2, SARS-CoV, and MERS-CoV genomes. Microbes Infect. 2020, 22, 226–229. [Google Scholar] [CrossRef]
- Narazaki, M.; Kishimoto, T. The Two-Faced Cytokine IL-6 in Host Defense and Diseases. Int. J. Mol. Sci. 2018, 19, 3528. [Google Scholar] [CrossRef] [Green Version]
- Anderluh, M.; Jug, G.; Svajger, U.; Obermajer, N. DC-SIGN antagonists, a potential new class of anti-infectives. Curr. Med. Chem. 2012, 19, 992–1007. [Google Scholar] [CrossRef]
- Khoo, U.S.; Chan, K.Y.; Chan, V.S.; Lin, C.L. DC-SIGN and L-SIGN: The SIGNs for infection. J. Mol. Med. 2008, 86, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.L.; Harman, A.N.; Donaghy, H. DC-SIGN ‘AIDS’ HIV immune evasion and infection. Nat. Immunol. 2007, 8, 556–558. [Google Scholar] [CrossRef]
- Jain, P.; Manuel, S.L.; Khan, Z.K.; Ahuja, J.; Quann, K.; Wigdahl, B. DC-SIGN mediates cell-free infection and transmission of human T-cell lymphotropic virus type 1 by dendritic cells. J. Virol. 2009, 83, 10908–10921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, L.; Roosendahl, P.; Ng, W.C.; Brooks, A.G.; Reading, P.C.; Londrigan, S.L. Endocytic function is critical for influenza A virus infection via DC-SIGN and L-SIGN. Sci. Rep. 2016, 6, 19428. [Google Scholar] [CrossRef]
- Kwon, D.S.; Gregorio, G.; Bitton, N.; Hendrickson, W.A.; Littman, D.R. DC-SIGN-mediated internalization of HIV is required for trans-enhancement of T cell infection. Immunity 2002, 16, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Pustylnikov, S.; Dave, R.S.; Khan, Z.K.; Porkolab, V.; Rashad, A.A.; Hutchinson, M.; Fieschi, F.; Chaiken, I.; Jain, P. Short Communication: Inhibition of DC-SIGN-Mediated HIV-1 Infection by Complementary Actions of Dendritic Cell Receptor Antagonists and Env-Targeting Virus Inactivators. AIDS Res. Hum. Retrovir. 2016, 32, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.Y.; Huang, Y.; Ganesh, L.; Leung, K.; Kong, W.P.; Schwartz, O.; Subbarao, K.; Nabel, G.J. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 2004, 78, 5642–5650. [Google Scholar] [CrossRef] [Green Version]
- Amraie, R.; Napoleon, M.A.; Yin, W.; Berrigan, J.; Suder, E.; Zhao, G.; Olejnik, J.; Gummuluru, S.; Muhlberger, E.; Chitalia, V.; et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2 and are differentially expressed in lung and kidney epithelial and endothelial cells. Biology 2020, 10, 1. [Google Scholar] [CrossRef]
- Nurden, A.T.; Nurden, P.; Sanchez, M.; Andia, I.; Anitua, E. Platelets and wound healing. Front. Biosci. 2008, 13, 3532–3548. [Google Scholar] [CrossRef]
- Tanaka, T.; Kishimoto, T. Targeting Interleukin-6: All the Way to Treat Autoimmune and Inflammatory Diseases. Int. J. Biol. Sci. 2012, 8, 1227–1236. [Google Scholar] [CrossRef]
- Yang, M.-L.; Wang, C.-T.; Yang, S.-J.; Leu, C.-H.; Chen, S.-H.; Wu, C.-L.; Shiau, A.-L. IL-6 ameliorates acute lung injury in influenza virus infection. Sci. Rep. 2017, 7, 43829. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlberg, J.; Chong, D.S.Y.; Lai, W.Y.Y. Do men have a higher case fatality rate of severe acute respiratory syndrome than women do? Am. J. Epidemiol. 2004, 159, 229–231. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Chughtai, A.A.; Dyda, A.; MacIntyre, C.R. Comparative epidemiology of Middle East respiratory syndrome coronavirus (MERS-CoV) in Saudi Arabia and South Korea. Emerg. Microbes Infect. 2017, 6, e51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, D.B.G.; Shah, A.; Doubeni, C.A.; Sia, I.G.; Wieland, M.L. The Disproportionate Impact of COVID-19 on Racial and Ethnic Minorities in the United States. Clin. Infect. Dis. 2020, 72, 703–706. [Google Scholar] [CrossRef]
- Rothan, H.A.; Acharya, A.; Reid, S.P.; Kumar, M.; Byrareddy, S.N. Molecular Aspects of COVID-19 Differential Pathogenesis. Pathogens 2020, 9, 538. [Google Scholar] [CrossRef]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.-C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Stepanikova, I.; Bateman, L.B.; Oates, G.R. Systemic Inflammation in Midlife: Race, Socioeconomic Status, and Perceived Discrimination. Am. J. Prev. Med. 2017, 52, S63–S76. [Google Scholar] [CrossRef] [Green Version]
- Singu, S.; Acharya, A.; Challagundla, K.; Byrareddy, S.N. Impact of Social Determinants of Health on the Emerging COVID-19 Pandemic in the United States. Front. Public Health 2020, 8, 406. [Google Scholar] [CrossRef]
- O’Donovan, A.; Neylan, T.C.; Metzler, T.; Cohen, B.E. Lifetime exposure to traumatic psychological stress is associated with elevated inflammation in the Heart and Soul Study. Brain Behav. Immun. 2012, 26, 642–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vepa, A.; Bae, J.P.; Ahmed, F.; Pareek, M.; Khunti, K. COVID-19 and ethnicity: A novel pathophysiological role for inflammation. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Holmes, L., Jr.; Enwere, M.; Williams, J.; Ogundele, B.; Chavan, P.; Piccoli, T.; Chinacherem, C.; Comeaux, C.; Pelaez, L.; Okundaye, O.; et al. Black-White Risk Differentials in COVID-19 (SARS-COV2) Transmission, Mortality and Case Fatality in the United States: Translational Epidemiologic Perspective and Challenges. Int. J. Environ. Res. Public Health 2020, 17, 4322. [Google Scholar] [CrossRef]
- Laurencin, C.T.; McClinton, A. The COVID-19 Pandemic: A Call to Action to Identify and Address Racial and Ethnic Disparities. J. Racial Ethn. Health Disparities 2020, 7, 398–402. [Google Scholar] [CrossRef]
- Spinola, H. HLA Loci and Respiratory Infectious Diseases. J. Respir. Res. 2016, 2, 56–66. [Google Scholar]
- Wang, W.; Zhang, W.; Zhang, J.; He, J.; Zhu, F. Distribution of HLA allele frequencies in 82 Chinese individuals with coronavirus disease-2019 (COVID-19). Hla 2020, 96, 194–196. [Google Scholar] [CrossRef]
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human leukocyte antigen susceptibility map for SARS-CoV-2. J. Virol. 2020, 94, e00510–e00520. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Cheng, G.; Chui, C.H.; Lau, F.Y.; Chan, P.K.S.; Ng, M.H.L.; Sung, J.J.Y.; Wong, R.S.M. ABO Blood Group and Susceptibility to Severe Acute Respiratory Syndrome. JAMA 2020, 293, 1447–1451. [Google Scholar] [CrossRef]
- Olwenyi, O.A.; Dyavar, S.R.; Acharya, A.; Podany, A.T.; Fletcher, C.V.; Ng, C.L.; Reid, S.P.; Byrareddy, S.N. Immuno-epidemiology and pathophysiology of coronavirus disease 2019 (COVID-19). J. Mol. Med. 2020, 98, 1369–1383. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Tapias, P.; Castiblanco, J.; Anaya, J.-M. Major histocompatibility complex: Antigen processing and presentation. In Autoimmunity: From Bench to Bedside; El Rosario University Press: Bogata, Columbia, 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459467/ (accessed on 15 August 2020).
- Reference, G.H. Histocompatibility Complex. Available online: https://www.ncbi.nlm.nih.gov/pubmed/ (accessed on 15 August 2020).
- Hilton, H.G.; McMurtrey, C.P.; Han, A.S.; Djaoud, Z.; Guethlein, L.A.; Blokhuis, J.H.; Pugh, J.L.; Goyos, A.; Horowitz, A.; Buchli, R.; et al. The Intergenic Recombinant HLA-B∗46:01 Has a Distinctive Peptidome that Includes KIR2DL3 Ligands. Cell Rep. 2017, 19, 1394–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Galarza, F.F.; McCabe, A.; Dos Santos, E.J.; Takeshita, L.; Ghattaoraya, G.; Jones, A.R.; Middleton, D. Allele Frequency Net Database (AFND). Available online: http://www.allelefrequencies.net/hla6006a.asp?hla_locus_type=Classical&hla_locus=B&hla_allele1=B*46%3A01&hla_allele2=B*46%3A01&hla_selection=&hla_pop_selection=&hla_population=&hla_country=&hla_dataset=&hla_region=&hla_ethnic=&hla_study=&hla_order=order_1&hla_sample_size_pattern=equal&hla_sample_size=&hla_sample_year_pattern=equal&hla_sample_year=&hla_level_pattern=equal&hla_level=&standard=a&hla_show= (accessed on 15 August 2020).
- Lin, M.; Tseng, H.-K.; Trejaut, J.A.; Lee, H.-L.; Loo, J.-H.; Chu, C.-C.; Chen, P.-J.; Su, Y.-W.; Lim, K.H.; Tsai, Z.-U.; et al. Association of HLA class I with severe acute respiratory syndrome coronavirus infection. BMC Med. Genet. 2003, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, M.H.L.; Lau, K.M.; Li, L.; Cheng, S.H.; Chan, W.Y.; Hui, P.K.; Zee, B.; Leung, C.B.; Sung, J.J.Y. Association of Human-Leukocyte-Antigen Class I (B*0703) and Class II (DRB1*0301) Genotypes with Susceptibility and Resistance to the Development of Severe Acute Respiratory Syndrome. J. Infect. Dis. 2004, 190, 515–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barquera, R.; Collen, E.; Di, D.; Buhler, S.; Teixeira, J.; Llamas, B.; Nunes, J.M.; Sanchez-Mazas, A. Binding affinities of 438 HLA proteins to complete proteomes of seven pandemic viruses and distributions of strongest and weakest HLA peptide binders in populations worldwide. Hla 2020, 96, 277–298. [Google Scholar] [CrossRef]
- Levin, A.M.; Adrianto, I.; Datta, I.; Iannuzzi, M.C.; Trudeau, S.; Li, J.; Drake, W.P.; Montgomery, C.G.; Rybicki, B.A. Association of HLA-DRB1 with Sarcoidosis Susceptibility and Progression in African Americans. Am. J. Respir. Cell Mol. Biol. 2015, 53, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDCgov. COVID-19 in Racial and Ethnic Minority Groups|CDC; U.S Department of Health and Human Services: Washington, DC, USA, 2021.
- Ganji, A.; Farahani, I.; Khansarinejad, B.; Ghazavi, A.; Mosayebi, G. Increased expression of CD8 marker on T-cells in COVID-19 patients. Blood Cells Mol. Dis. 2020, 83, 102437. [Google Scholar] [CrossRef]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Kitamura, H.; Takahashi, N.; Ohtake, J.; Kaneumi, S.; Sumida, K.; Homma, S.; Kawamura, H.; Minagawa, N.; Shibasaki, S.; et al. IL-6 down-regulates HLA class II expression and IL-12 production of human dendritic cells to impair activation of antigen-specific CD4(+) T cells. Cancer Immunol. Immunother. 2016, 65, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assiri, A.; Al-Tawfiq, J.A.; Al-Rabeeah, A.A.; Al-Rabiah, F.A.; Al-Hajjar, S.; Al-Barrak, A.; Flemban, H.; Al-Nassir, W.N.; Balkhy, H.H.; Al-Hakeem, R.F.; et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: A descriptive study. Lancet. Infect. Dis. 2013, 13, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Monneret, G.; Lepape, A.; Voirin, N.; Bohé, J.; Venet, F.; Debard, A.-L.; Thizy, H.; Bienvenu, J.; Gueyffier, F.; Vanhems, P. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med. 2006, 32, 1175–1183. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, Y.; Huang, H.; Li, D.; Gu, D.; Lu, X.; Zhang, Z.; Liu, L.; Liu, T.; Liu, Y.; et al. Relationship between the ABO Blood Group and the COVID-19 Susceptibility. Clin. Infect. Dis. 2020, 73, 328–331. [Google Scholar] [CrossRef]
- Zietz, M.; Tatonetti, N.P. Testing the association between blood type and COVID-19 infection, intubation, and death. MedRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Latz, C.A.; DeCarlo, C.; Boitano, L.; Png, C.M.; Patell, R.; Conrad, M.F.; Eagleton, M.; Dua, A. Blood type and outcomes in patients with COVID-19. Ann. Hematol. 2020, 2113–2118. [Google Scholar] [CrossRef]
- Preece, A.F.; Strahan, K.M.; Devitt, J.; Yamamoto FI Gustafsson, K. Expression of ABO or related antigenic carbohydrates on viral envelopes leads to neutralization in the presence of serum containing specific natural antibodies and complement. Blood 2002, 99. [Google Scholar] [CrossRef] [Green Version]
- Guillon, P.; Clément, M.; Sébille, V.; Rivain, J.G.; Chou, C.F.; Ruvoën-Clouet, N.; Le Pendu, J. Inhibition of the interaction between the SARS-CoV Spike protein and its cellular receptor by anti-histo-blood group antibodies. Glycobiology 2008, 18, 1085–1093. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.J.; Busse, W.W.; Bacharier, L.B.; Kattan, M.; O’Connor, G.T.; Wood, R.A.; Visness, C.M.; Durham, S.R.; Larson, D.; Esnault, S.; et al. Association of respiratory allergy, asthma, and expression of the SARS-CoV-2 receptor ACE2. J. Allergy Clin. Immunol. 2020, 146, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Farsalinos, K.; Niaura, R.; Le Houezec, J.; Barbouni, A.; Tsatsakis, A.; Kouretas, D.; Vantarakis, A.; Poulas, K. Editorial: Nicotine and SARS-CoV-2: COVID-19 may be a disease of the nicotinic cholinergic system. Toxicol. Rep. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dong, Y.Q.; Yin, J.; Yao, J.; Shen, J.; Sheng, G.J.; Li, K.; Lv, H.F.; Fang, X.; Wu, W.F. Meta-analysis of vitamin D and lung function in patients with asthma. Respir. Res. 2019, 20, 161. [Google Scholar] [CrossRef]
- Aranow, C. Vitamin D and the immune system. J. Investig. Med. 2011, 59, 881–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, W.B.; Lahore, H.; McDonnell, S.L.; Baggerly, C.A.; French, C.B.; Aliano, J.L.; Bhattoa, H.P. Evidence that Vitamin D Supplementation Could Reduce Risk of Influenza and COVID-19 Infections and Deaths. Nutrients 2020, 12, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilie, P.C.; Stefanescu, S.; Smith, L. The role of vitamin D in the prevention of coronavirus disease 2019 infection and mortality. Aging Clin. Exp. Res. 2020, 32, 1195–1198. [Google Scholar] [CrossRef]
- McMullan, C.J.; Borgi, L.; Curhan, G.C.; Fisher, N.; Forman, J.P. The effect of vitamin D on renin-angiotensin-system activation and blood pressure-a randomized control trial. J. Hypertens. 2017, 35, 822. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, F. Vitamin-D and COVID-19: Do deficient risk a poorer outcome? Lancet Diabetes Endocrinol. 2020, 8, 570. [Google Scholar] [CrossRef]
- Zisi, D.; Challa, A.; Makis, A. The association between vitamin D status and infectious diseases of the respiratory system in infancy and childhood. Hormones 2019, 18, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Dankers, W.; Colin, E.M.; van Hamburg, J.P.; Lubberts, E. Vitamin D in Autoimmunity: Molecular Mechanisms and Therapeutic Potential. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherberich, J.E.; Kellermeyer, M.; Ried, C.; Hartinger, A. 1-alpha-calcidol modulates major human monocyte antigens and toll-like receptors TLR 2 and TLR4 in vitro. Eur. J. Med. Res. 2005, 10, 179–182. [Google Scholar]
- Brosbøl-Ravnborg, A.; Bundgaard, B.; Höllsberg, P. Synergy between Vitamin D3 and Toll-Like Receptor Agonists Regulates Human Dendritic Cell Response during Maturation. Clin. Dev. Immunol. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Aygun, H. Vitamin D can prevent COVID-19 infection-induced multiple organ damage. In Naunyn-Schmiedeberg’s Archives of Pharmacology Naunyn-Schmiedeberg’s Archives of Pharmacology; Springer: Heidelberg, Germany, 2020; pp. 1–4. [Google Scholar] [CrossRef]
- Herold, T.; Jurinovic, V.; Arnreich, C.; Lipworth, B.J.; Hellmuth, J.C.; von Bergwelt-Baildon, M.; Klein, M.; Weinberger, T. Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19. J. Allergy Clin. Immunol. 2020, 146, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M.; et al. High Prevalence of Obesity in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Requiring Invasive Mechanical Ventilation. Obesity 2020, 28, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Wortsman, J.; Matsuoka, L.Y.; Chen, T.C.; Lu, Z.; Holick, M.F. Decreased bioavailability of vitamin D in obesity. Am. J. Clin. Nutr. 2000, 72, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.S.; Azhar, E.I.; Kim, Y.J.; Memish, Z.A.; Oh, M.D.; Zumla, A. Middle East respiratory syndrome coronavirus: Risk factors and determinants of primary, household, and nosocomial transmission. Lancet Infect. Dis. 2018, 18, e217–e227. [Google Scholar] [CrossRef] [Green Version]
- Amaral, W.Z.; Krueger, R.F.; Ryff, C.D.; Coe, C.L. Genetic and environmental determinants of population variation in interleukin-6, its soluble receptor and C-reactive protein: Insights from identical and fraternal twins. Brain Behav. Immun. 2015, 49, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Lange, N.E.; Sparrow, D.; Vokonas, P.; Litonjua, A.A. Vitamin D deficiency, smoking, and lung function in the Normative Aging Study. Am. J. Respir. Crit. Care Med. 2012, 186, 616–621. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, A.G.; Lin, M.; Wu, J.; Kennedy, T.P.; Daley, L.-A.; Ashby, C.R., Jr.; Mantell, L.L. From nicotine to the cholinergic anti-inflammatory reflex–Can nicotine alleviate the dysregulated inflammation in COVID-19? J. Immunotoxicol. 2021, 18, 23–29. [Google Scholar] [CrossRef] [PubMed]
- White, S.R.; Laxman, B.; Naureckas, E.T.; Hogarth, D.K.; Solway, J.; Sperling, A.I.; Ober, C. Evidence for an IL-6-high asthma phenotype in asthmatic patients of African ancestry. J. Allergy Clin. Immunol. 2019, 144, 304–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Hasegawa, K.; Ma, B.; Fujiogi, M.; Camargo, C.; Liang, L. Association of asthma and its genetic predisposition with the risk of severe COVID-19. J. Allergy Clin. Immunol. 2020, 142, 327–329. [Google Scholar] [CrossRef] [PubMed]
- van Paassen, J.; Vos, J.S.; Hoekstra, E.M.; Neumann, K.M.; Boot, P.C.; Arbous, S.M. Corticosteroid use in COVID-19 patients: A systematic review and meta-analysis on clinical outcomes. Crit. Care 2020, 24, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Della-Torre, E.; Campochiaro, C.; Cavalli, G.; De Luca, G.; Napolitano, A.; La Marca, S.; Boffini, N.; Da Prat, V.; Di Terlizzi, G.; Lanzillotta, M.; et al. Interleukin-6 blockade with sarilumab in severe COVID-19 pneumonia with systemic hyperinflammation: An open-label cohort study. Ann. Rheum. Dis. 2020, 79, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, V.; Shannon, C.P.; Wei, X.S.; Xiang, X.; Wang, X.; Wang, Z.H.; Tebbutt, S.J.; Kollmann, T.R.; Fish, E.N. Interferon-alpha2b Treatment for COVID-19. Front. Immunol. 2020, 11, 1061. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Voell, J.; Kumar, P.; Raviprakash, K.; Wu, H.; Jiao, J.A.; Sullivan, E.; Luke, T.; Davey, R.T., Jr. Safety and tolerability of a novel, polyclonal human anti-MERS coronavirus antibody produced from transchromosomic cattle: A phase 1 randomised, double-blind, single-dose-escalation study. Lancet Infect. Dis. 2018, 18, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.H.; Acharya, A.; Mohan, M.; Byrareddy, S.N. COVID-19 and AIDS: Outcomes from the coexistence of two global pandemics and the importance of chronic antiretroviral therapy. J. Med. Virol. 2021, 93, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.T.; Qian, J.D.; Zhu, W.Y.; Wang, Y.; Wang, G.Q. A systematic review of lopinavir therapy for SARS coronavirus and MERS coronavirus-A possible reference for coronavirus disease-19 treatment option. J. Med. Virol. 2020, 92, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Folegatti, P.M.; Bittaye, M.; Flaxman, A.; Lopez, F.R.; Bellamy, D.; Kupke, A.; Mair, C.; Makinson, R.; Sheridan, J.; Rohde, C.; et al. Safety and immunogenicity of a candidate Middle East respiratory syndrome coronavirus viral-vectored vaccine: A dose-escalation, open-label, non-randomised, uncontrolled, phase 1 trial. Lancet Infect. Dis. 2020, 20, 816–826. [Google Scholar] [CrossRef]
- Plaze, M.; Attali, D.; Petit, A.C.; Blatzer, M.; Simon-Loriere, E.; Vinckier, F.; Cachia, A.; Chretien, F.; Gaillard, R. Repurposing chlorpromazine to treat COVID-19: The reCoVery study. Encephale 2020, 46, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.D.; Shukla, S.; Chung, Y.H.; Beiss, V.; Chan, S.K.; Ortega-Rivera, O.A.; Wirth, D.M.; Chen, A.; Sack, M.; Pokorski, J.K.; et al. COVID-19 vaccine development and a potential nanomaterial path forward. Nat. Nanotechnol. 2020, 15, 646–655. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. COVID Data Tracker; U.S. Department of Health and Human Services, Ed.; Centers for Disease Control and Prevention: Washington, DC, USA, 2021.
- Pang, J.; Wang, M.X.; Ang, I.Y.H.; Tan, S.H.X.; Lewis, R.F.; Chen, J.I.; Gutierrez, R.A.; Gwee, S.X.W.; Chua, P.E.Y.; Yang, Q.; et al. Potential Rapid Diagnostics, Vaccine and Therapeutics for 2019 Novel Coronavirus (2019-nCoV): A Systematic Review. J. Clin. Med. 2020, 9, 623. [Google Scholar] [CrossRef] [Green Version]
- Tse, L.V.; Meganck, R.M.; Graham, R.L.; Baric, R.S. The Current and Future State of Vaccines, Antivirals and Gene Therapies Against Emerging Coronaviruses. Front. Microbiol. 2020, 11, 658. [Google Scholar] [CrossRef] [PubMed]
- Koch, T.; Dahlke, C.; Fathi, A.; Kupke, A.; Krahling, V.; Okba, N.M.A.; Halwe, S.; Rohde, C.; Eickmann, M.; Volz, A.; et al. Safety and immunogenicity of a modified vaccinia virus Ankara vector vaccine candidate for Middle East respiratory syndrome: An open-label, phase 1 trial. Lancet Infect. Dis. 2020, 20, 827–838. [Google Scholar] [CrossRef]
- Rodriguez-Diaz, C.E.; Guilamo-Ramos, V.; Mena, L.; Hall, E.; Honermann, B.; Crowley, J.S.; Baral, S.; Prado, G.J.; Marzan-Rodriguez, M.; Beyrer, C.; et al. Risk for COVID-19 infection and death among Latinos in the United States: Examining heterogeneity in transmission dynamics. Ann. Epidemiol. 2020. [Google Scholar] [CrossRef]
- Spratt, A.N.; Kannan, S.R.; Woods, L.T.; Weisman, G.A.; Quinn, T.P.; Lorson, C.L.; Sonnerborg, A.; Byrareddy, S.N.; Singh, K. Factors Associated with Emerging and Re-emerging of SARS-CoV-2 Variants. BioRxiv 2021. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, R.; Joshi, S.R.; Misra, A. Mucormycosis in COVID-19: A systematic review of cases reported worldwide and in India. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S. Convergent evolution of SARS-CoV-2 spike mutations, L452R, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. BioRxiv 2021, 9, 1542. [Google Scholar]
- Raut, A.; Huy, N.T. Rising incidence of mucormycosis in patients with COVID-19: Another challenge for India amidst the second wave? Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Fungal Diseases: Mucormycosis. Available online: https://www.cdc.gov/fungal/diseases/mucormycosis/index.html (accessed on 15 August 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Increased Risk Allele | Decreased Risk Allele |
|---|---|---|
| MERS | HLA-DRB1*11:01 HLA-DQB1*02:02 | |
| SARS-CoV-1 | HLA-B*46:01 HLA-B*5401 HLA-B*0703 HLA-DRB1*03:01 HLA-DRB1*12:02 | HLA-DR0301 |
| SARS-CoV-2 | HLA-B*46:01 HLA-DRB1*01:01 HLA-BRB1*03:02 HLA-BRB1*03:03 HLA-A*25:01 HLA-C*01:02 HLA-C*07:29 HLA-B*15:27 | DRB1*10:10 DRB1*01:01 DRB1*01:04 HLA-B*15:03 HLA-A*02:02 HLA-B*15:03 HLA-C:12:03 |
| Name | MOA | Clinical Outcome(s) | Comments | Trial |
|---|---|---|---|---|
| COVID-19 | ||||
| Tocilizumab | Humanized monoclonal IL-6R antibody | Decreased patient temperature, oxygen intake, and CRP levels | Currently used to treat rheumatoid arthritis | Phase 2/3 |
| Hydroxychloroquine | Unknown | Not significantly more effective than standard of care | Used to treat malaria and rheumatoid arthritis | Phase 3 |
| Favipiravir | Viral RdRp inhibitor | Increased viral clearance and improved chest CT scans | Effective against Ebola virus and influenza | Phase 2/3 |
| LPV/RTV | LPV: Protease inhibitor RTV: LPV metabolism inhibitor | Eliminated detectable SARS-CoV-2 viral titers (44% of patients) | No detectable side effects found | Phase 2 |
| Arbidol | Viral entry inhibitor | Eliminated detectable SARS-CoV-2 viral titers Delayed the progression of pulmonary lesions | Currently used to treat prophylaxis and influenza in Russia and China | Phase 4 |
| SARS | ||||
| Remdesivir | Viral RdRp inhibitor | Reduces pulmonary SARS-CoV viral load | Broad-spectrum antiviral | Preclinical |
| LPV/RTV | LPV: Protease inhibitor RTV: LPV metabolism inhibitor | Reduces SARS-CoV viral load, mortality, and intubation rate | Possibly most effective as an initial therapy against infection | Preclinical |
| MERS | ||||
| REGN 3048 and REGN 3051 | Humanized mAbs | Clinical trial results not yet published | None | Phase 1 |
| LPV/RTV and IFN-beta-1b | LPV/RTV: Protease inhibitors IFN-beta-1b: Antiviral protein activator | Contributed to a 40% decrease in early stage infection risk | None | Phase 1 |
| SAB-301 | Human polyclonal IgG | Clinical trial results not yet published | No reported tolerance issues | Phase 1 |
| Name | Developer(s) | Vaccine Type | Trial Phase |
|---|---|---|---|
| * mRNA 1273 | Moderna and NIAID | mRNA vaccine | Phase 3 |
| * ChAdOx1 | University of Oxford and AstraZeneca | Non-replicating viral vector | Phase 2b/3 |
| CoronaVacc | Sinovac | Inactivated vaccine | Phase 3 |
| N/A | Beijing Institute of Biological Products and Sinopharm | Inactivated vaccine | Phase 1/2 |
| N/A | Wuhan Institute of Biological Products and Sinopharm | Inactivated vaccine | Phase 1/2 |
| N/A | Institute of Medical Biology, Chinese Academy of Medical Sciences | Inactivated vaccine | Phase 1 |
| N/A | Novavax | Subunit vaccine | Phase 1/2 |
| Ad5-nCoV | CanSino Biological Incorporation, Beijing Institute of Biotechnology and Canadian Center for Vaccinology | Non-replicating viral vector (adenovirus type 5) | Phase 1 and phase 2 |
| N/A | Shenzhen Geno-Immune Medical Institute | Non-replicating viral vector | Procedure 1: Phase 1/2Procedure 2: Phase 1 |
| INO-4800 | Inovio Pharmaceuticals | DNA vaccine | Phase 1 |
| N/A | Symvivo | DNA vaccine | Phase 1 |
| * BNT162 | BioNTech, Pfizer, and Fosun Pharma | RNA vaccine | Phase 1/2 |
| * COVAXIN | Bharat Biotech | Inactivated vaccine | Phase 3 |
| Name | Developer | Vaccine Type | Clinical Outcome(s) | Comments | Trial Phase |
|---|---|---|---|---|---|
| SARS-CoV | |||||
| N/A | Sinovac | Inactivated vaccine | Contributed to 100% seroconversion following 2 vaccine doses of 16 SU | Short-term systemic adverse events (all subjects) | Phase 1 |
| N/A | NIH, NIAID (Vaccine Research Center) | Recombinant DNA vaccine | CD4+ T-cell response (All subjects) CD8+ T-cell response (20% of subjects) | Mild systemic reactions observed (~50% of subjects) | Phase 1 |
| MERS-CoV | |||||
| GLS 5300 | Inovio Pharmaceuticals, Inc. | DNA vaccine | Associated with seroconversion and nAb development against MERS-COV viral infection | Immune protection lasted for one year after vaccination | Phase 1 |
| ChAdOx1 MERS | Oxford University | ChAdOx1 vector | Associated with immune protection, seroconversion, and T-cell response | Immune protection lasted for a year after vaccination | Phase 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanna, J.; Tipparaju, P.; Mulherkar, T.; Lin, E.; Mischley, V.; Kulkarni, R.; Bolton, A.; Byrareddy, S.N.; Jain, P. Risk Factors Associated with the Clinical Outcomes of COVID-19 and Its Variants in the Context of Cytokine Storm and Therapeutics/Vaccine Development Challenges. Vaccines 2021, 9, 938. https://doi.org/10.3390/vaccines9080938
Hanna J, Tipparaju P, Mulherkar T, Lin E, Mischley V, Kulkarni R, Bolton A, Byrareddy SN, Jain P. Risk Factors Associated with the Clinical Outcomes of COVID-19 and Its Variants in the Context of Cytokine Storm and Therapeutics/Vaccine Development Challenges. Vaccines. 2021; 9(8):938. https://doi.org/10.3390/vaccines9080938
Chicago/Turabian StyleHanna, John, Padmavathi Tipparaju, Tania Mulherkar, Edward Lin, Victoria Mischley, Ratuja Kulkarni, Aliyah Bolton, Siddappa N. Byrareddy, and Pooja Jain. 2021. "Risk Factors Associated with the Clinical Outcomes of COVID-19 and Its Variants in the Context of Cytokine Storm and Therapeutics/Vaccine Development Challenges" Vaccines 9, no. 8: 938. https://doi.org/10.3390/vaccines9080938