
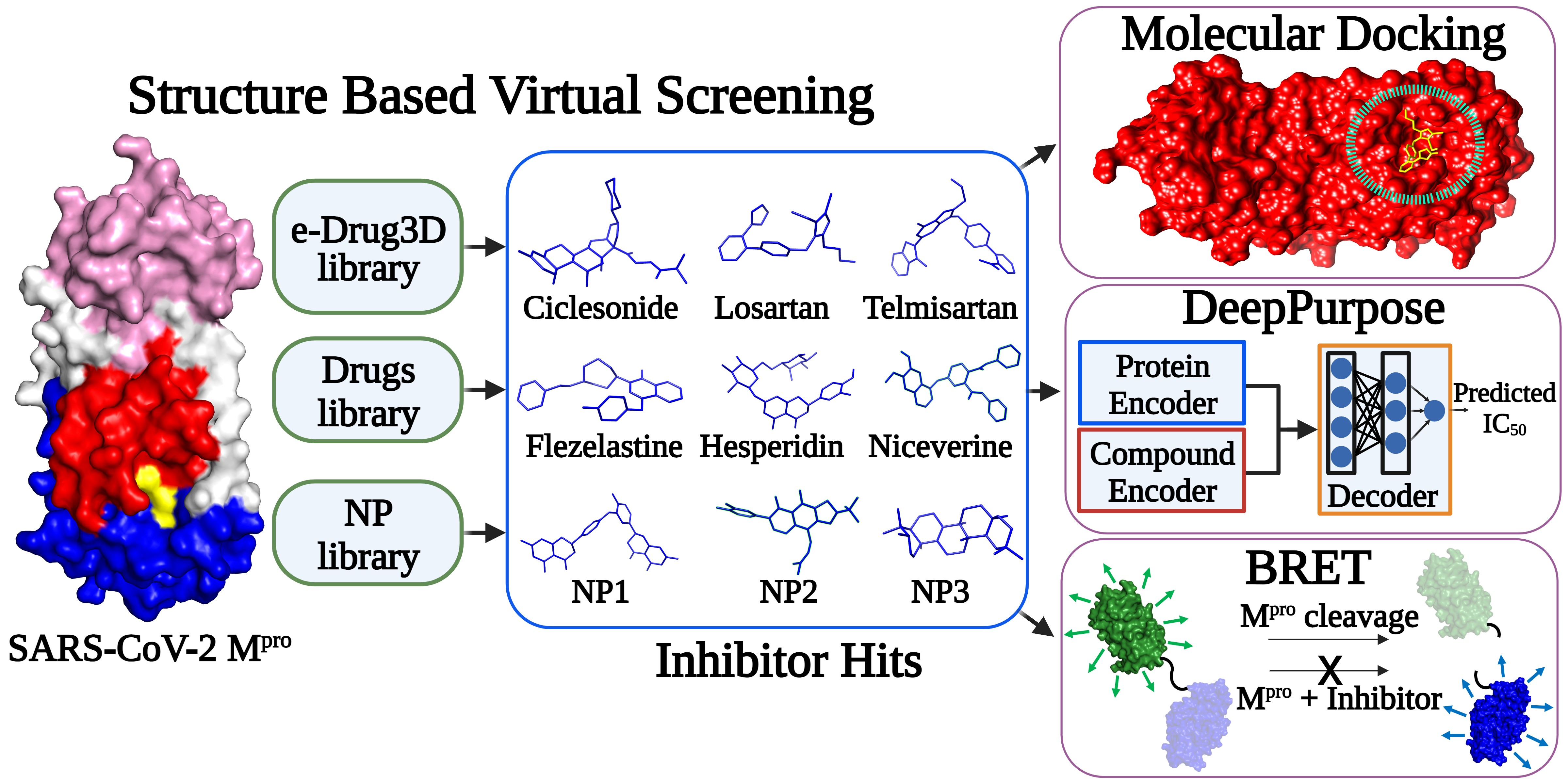
Structure-Based Virtual Screening and Functional Validation of Potential Hit Molecules Targeting the SARS-CoV-2 Main Protease

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Retrieval and Preparation of Mpro for Virtual Screening
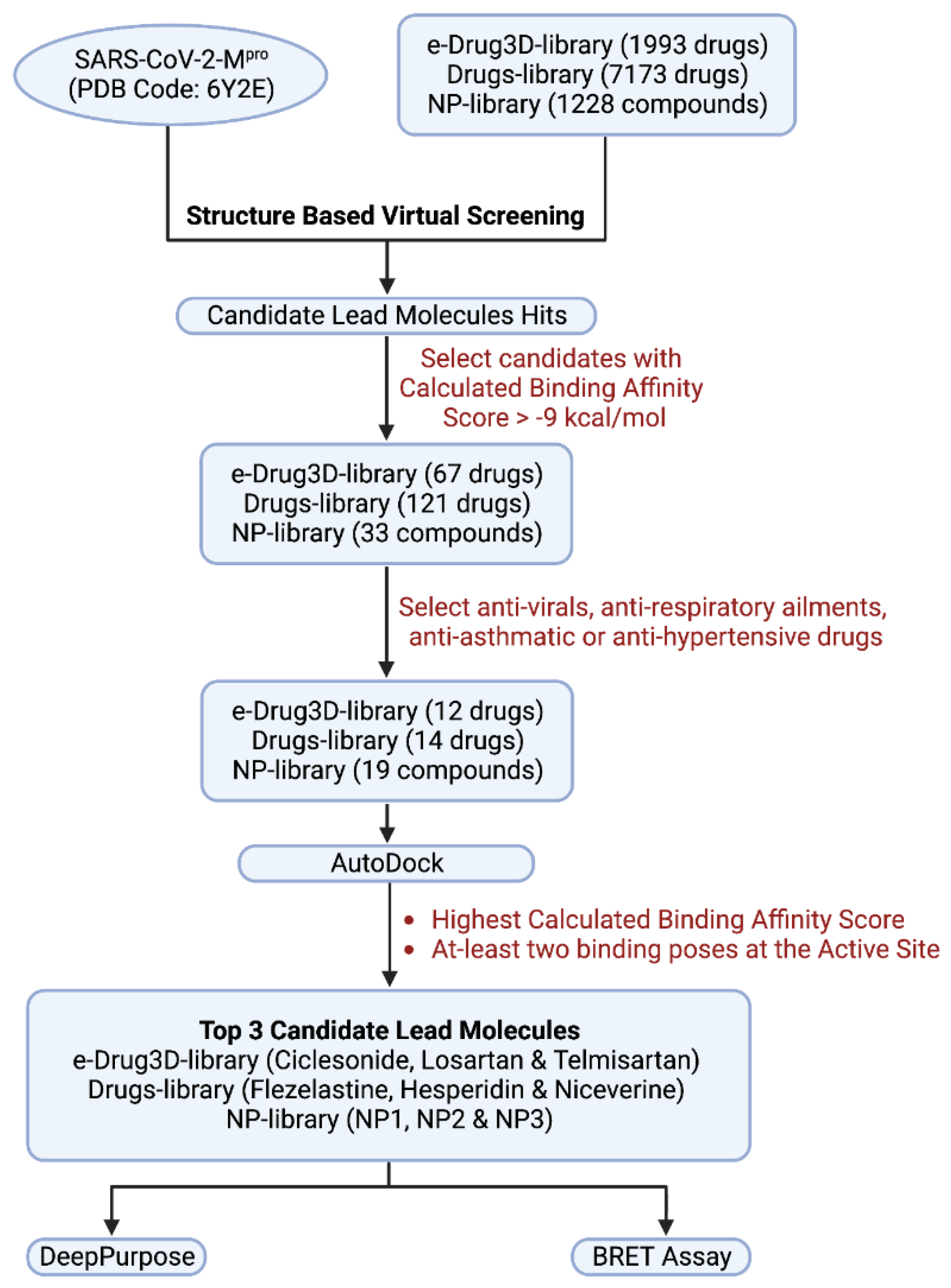
2.2. Library Selection
2.3. Ligand Extraction and Preparation
2.4. Docking Parameters
2.5. Virtual Drug Screening through Molecular Docking Studies
2.6. Analysis and Visualization of Mpro-Drug Complex
2.7. Drug Purchase Information
2.8. Prediction of Mpro-Hit Molecule Interactions Using Deep Learning
2.9. Cell Lysate Preparation for In Vitro BRET Assay
2.10. Expression and Purification of Recombinant SARS-CoV-2 Mpro
2.11. In Vitro BRET-Based Mpro Proteolytic Cleavage Inhibitor Assay
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, E.; Du, H.; Gardner, L. An Interactive Web-Based Dashboard to Track COVID-19 in Real Time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Tang, B.; Bragazzi, N.L.; Li, Q.; Tang, S.; Xiao, Y.; Wu, J. An Updated Estimation of the Risk of Transmission of the Novel Coronavirus (2019-NCov). Infect. Dis. Model. 2020, 5, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.K.K.; Kausar, M.; Bisht, D.; Kumar, D.; Sati, D.; Rajpal, G. Recent Diagnostic Techniques for COVID-19 BT—Computational Intelligence Techniques for Combating COVID-19; Kautish, S., Peng, S.-L., Obaid, A.J., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 75–94. ISBN 978-3-030-68936-0. [Google Scholar]
- Ashour, H.M.; Elkhatib, W.F.; Rahman, M.; Elshabrawy, H.A. Insights into the Recent 2019 Novel Coronavirus (SARS-CoV-2) in Light of Past Human Coronavirus Outbreaks. Pathogens 2020, 9, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 Coronavirus Structure, Mechanism of Action, Antiviral Drug Promises and Rule out against Its Treatment. J. Biomol. Struct. Dyn. 2021, 39, 3409–3418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved $α$-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Muralidharan, N.; Sakthivel, R.; Velmurugan, D.; Gromiha, M.M. Computational Studies of Drug Repurposing and Synergism of Lopinavir, Oseltamivir and Ritonavir Binding with SARS-CoV-2 Protease against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 2673–2678. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yiu, C.-P.B.; Wong, K.-Y. Prediction of the SARS-CoV-2 (2019-NCoV) 3C-like Protease (3CL pro) Structure: Virtual Screening Reveals Velpatasvir, Ledipasvir, and Other Drug Repurposing Candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, R.J.; Jha, R.K.; Amera, G.M.; Jain, M.; Singh, E.; Pathak, A.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. Targeting SARS-CoV-2: A Systematic Drug Repurposing Approach to Identify Promising Inhibitors against 3C-like Proteinase and 2′-O-Ribose Methyltransferase. J. Biomol. Struct. Dyn. 2021, 39, 2679–2692. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Li, X.; Yu, Z.; Wu, S.; Ding, L.; Liu, J. Identification of Lactoferrin-Derived Peptides as Potential Inhibitors against the Main Protease of SARS-CoV-2. LWT 2022, 154, 112684. [Google Scholar] [CrossRef]
- Gambacorta, N.; Caputo, L.; Quintieri, L.; Monaci, L.; Ciriaco, F.; Nicolotti, O. Rational Discovery of Antiviral Whey Protein-Derived Small Peptides Targeting the SARS-CoV-2 Main Protease. Biomedicines 2022, 10, 1067. [Google Scholar] [CrossRef]
- Pandey, A.K.; Verma, S. An In-Silico Evaluation of Dietary Components for Structural Inhibition of SARS-Cov-2 Main Protease. J. Biomol. Struct. Dyn. 2022, 40, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.M.; Rey, J.; Lagorce, D.; Vavruša, M.; Becot, J.; Sperandio, O.; Villoutreix, B.O.; Tufféry, P.; Miteva, M.A. MTiOpenScreen: A Web Server for Structure-Based Virtual Screening. Nucleic Acids Res. 2015, 43, W448–W454. [Google Scholar] [CrossRef] [Green Version]
- Pihan, E.; Colliandre, L.; Guichou, J.-F.; Douguet, D. E-Drug3D: 3D Structure Collections Dedicated to Drug Repurposing and Fragment-Based Drug Design. Bioinformatics 2012, 28, 1540–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fu, T.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. DeepPurpose: A Deep Learning Library for Drug--Target Interaction Prediction. Bioinformatics 2020, 36, 5545–5547. [Google Scholar] [CrossRef] [PubMed]
- Geethakumari, A.M.; Ahmed, W.S.; Rasool, S.; Fatima, A.; Nasir Uddin, S.M.; Aouida, M.; Biswas, K.H. A Genetically Encoded BRET-Based SARS-CoV-2 Mpro Protease Activity Sensor. Commun. Chem. 2022, 5, s42004–s42022. [Google Scholar] [CrossRef]
- Grum-Tokars, V.; Ratia, K.; Begaye, A.; Baker, S.C.; Mesecar, A.D. Evaluating the 3C-like Protease Activity of SARS-Coronavirus: Recommendations for Standardized Assays for Drug Discovery. Virus Res. 2008, 133, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Panday, S.K.; Ghosh, I. In Silico Structure-Based Prediction of Receptor—Ligand Binding Affinity: Current Progress and Challenges. Struct. Bioinform. Appl. Preclin. Drug Discov. Process 2019, 27, 109–175. [Google Scholar]
- Azam, S.S.; Abbasi, S.W. Molecular Docking Studies for the Identification of Novel Melatoninergic Inhibitors for Acetylserotonin-O-Methyltransferase Using Different Docking Routines. Theor. Biol. Med. Model 2013, 10, 63. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Perez-Sanchez, H.; Lightstone, F.C. A Comprehensive Docking and MM/GBSA Rescoring Study of Ligand Recognition upon Binding Antithrombin. Curr. Top. Med. Chem. 2017, 17, 1631–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altamash, T.; Ahmed, W.; Rasool, S.; Biswas, K.H. Intracellular Ionic Strength Sensing Using NanoLuc. Int. J. Mol. Sci. 2021, 22, 677. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.H.; Badireddy, S.; Rajendran, A.; Anand, G.S.; Visweswariah, S.S. Cyclic Nucleotide Binding and Structural Changes in the Isolated GAF Domain of Anabaena Adenylyl Cyclase, CyaB2. PeerJ 2015, 3, e882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, K.H.; Visweswariah, S.S. Distinct Allostery Induced in the Cyclic GMP-Binding, Cyclic GMP-Specific Phosphodiesterase (PDE5) by Cyclic GMP, Sildenafil, and Metal Ions. J. Biol. Chem. 2011, 286, 8545–8554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, K.H.; Sopory, S.; Visweswariah, S.S. The GAF Domain of the CGMP-Binding, CGMP-Specific Phosphodiesterase (PDE5) Is a Sensor and a Sink for CGMP. Biochemistry 2008, 47, 3534–3543. [Google Scholar] [CrossRef] [PubMed]
- den Hamer, A.; Dierickx, P.; Arts, R.; de Vries, J.S.P.M.; Brunsveld, L.; Merkx, M. Bright Bioluminescent BRET Sensor Proteins for Measuring Intracellular Caspase Activity. ACS Sens. 2017, 2, 729–734. [Google Scholar] [CrossRef] [Green Version]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massagué, A.; Garcia-Vallvé, S.; Pujadas, G. Haste Makes Waste: A Critical Review of Docking-based Virtual Screening in Drug Repurposing for SARS-CoV-2 Main Protease (M-pro) Inhibition. Med. Res. Rev. 2022, 42, 744–769. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, K.; Yoshie, K.; Kurakami, Y.; Takahashi, K.; Kato, Y.; Morishima, T. Therapeutic Potential of Ciclesonide Inhalation for COVID-19 Pneumonia: Report of Three Cases. J. Infect. Chemother. 2020, 26, 625–632. [Google Scholar] [CrossRef]
- Matsuyama, S.; Kawase, M.; Nao, N.; Shirato, K.; Ujike, M.; Kamitani, W.; Shimojima, M.; Fukushi, S. The Inhaled Steroid Ciclesonide Blocks SARS-CoV-2 RNA Replication by Targeting the Viral Replication-Transcription Complex in Cultured Cells. J. Virol. 2020, 95, e01648-20. [Google Scholar] [CrossRef]
- Tsuchida, T.; Yamasaki, Y.; Kunishima, H.; Sato, K.; Kanazawa, M.; Moriuchi, A.; Morikawa, D.; Takita, M.; Naito, Y.; Fujii, S.; et al. Treatment of Two Cases of COVID-19 with Ciclesonide Resulted in Amelioration of Pneumonia Symptoms. Jpn. J. Antibiot. 2020, 73, 2. [Google Scholar]
- Nakajima, K.; Ogawa, F.; Sakai, K.; Uchiyama, M.; Oyama, Y.; Kato, H.; Takeuchi, I. A Case of Coronavirus Disease 2019 Treated with Ciclesonide. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2020; Volume 95, pp. 1296–1297. [Google Scholar]
- Nejat, R.; Sadr, A.S.; Freitas, B.; Crabttree, J.; Pegan, S.D.; Tripp, R.A.; Najafi, D. Losartan Inhibits SARS-CoV-2 Replication in Vitro: Losartan Promotes Cell Survival Following SARS-CoV-2 Infection in Vitro. J. Pharm. Pharm. Sci. 2021, 24, 390–399. [Google Scholar] [CrossRef]
- Rothlin, R.P.; Vetulli, H.M.; Duarte, M.; Pelorosso, F.G. Telmisartan as Tentative Angiotensin Receptor Blocker Therapeutic for COVID-19. Drug Dev. Res. 2020, 81, 768–770. [Google Scholar] [CrossRef]
- Yan, F.; Huang, F.; Xu, J.; Yang, P.; Qin, Y.; Lv, J.; Zhang, S.; Ye, L.; Gong, M.; Liu, Z.; et al. Antihypertensive Drugs Are Associated with Reduced Fatal Outcomes and Improved Clinical Characteristics in Elderly COVID-19 Patients. Cell Discov. 2020, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Puskarich, M.A.; Ingraham, N.E.; Merck, L.H.; Driver, B.E.; Wacker, D.A.; Black, L.P.; Jones, A.E.; Fletcher, C.V.; South, A.M.; Murray, T.A. Efficacy of Losartan in Hospitalized Patients With COVID-19–Induced Lung Injury: A Randomized Clinical Trial. JAMA Netw. Open 2022, 5, e222735. [Google Scholar] [CrossRef] [PubMed]
- Mirjalili, M.; Soodejani, M.T.; Raadabadi, M.; Dehghani, A.; Salemi, F. Does Losartan Reduce the Severity of COVID-19 in Hypertensive Patients? BMC Cardiovasc. Disord. 2022, 22, 116. [Google Scholar] [CrossRef] [PubMed]
- Reznikov, L.R.; Norris, M.H.; Vashisht, R.; Bluhm, A.P.; Li, D.; Liao, Y.-S.J.; Brown, A.; Butte, A.J.; Ostrov, D.A. Identification of Antiviral Antihistamines for COVID-19 Repurposing. Biochem. Biophys. Res. Commun. 2021, 538, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Laura, S.; Jérémie, B.; Jérôme, D.; Bérangère, B.; Clémentine, V.; François, C. Antihypertensive Drugs and COVID-19 Risk. Hypertension 2021, 77, 833–842. [Google Scholar]
- Bellavite, P.; Donzelli, A. Hesperidin and SARS-CoV-2: New Light on the Healthy Function of Citrus Fruits. Antioxidants 2020, 9, 742. [Google Scholar] [CrossRef] [PubMed]
- Ariyasena, J.; Baek, S.-H.; Perry, N.B.; Weavers, R.T. Ether-Linked Biflavonoids from Quintinia Acutifolia. J. Nat. Prod. 2004, 67, 693–696. [Google Scholar] [CrossRef]
- Ito, C.; Murata, T.; Itoigawa, M.; Nakao, K.; Kumagai, M.; Kaneda, N.; Furukawa, H. Induction of Apoptosis by Isoflavonoids from the Leaves of Millettia Taiwaniana in Human Leukemia HL-60 Cells. Planta. Med. 2006, 72, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Bell, E.W.; Yin, M.; Zhang, Y. EDock: Blind Protein–Ligand Docking by Replica-Exchange Monte Carlo Simulation. J. Cheminform. 2020, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Hou, N.; Peng, C.; Zhang, L.; Zhu, Y.; Hu, Q. BRET-Based Self-Cleaving Biosensors for SARS-CoV-2 3CLpro Inhibitor Discovery. Microbiol. Spectr. 2022, 10, e02559-21. [Google Scholar] [CrossRef]
- Ma, L.; Li, Q.; Xie, Y.; Yi, D.; Guo, S.; Guo, F.; Wang, J.; Yang, L.; Cen, S. Repurposing of HIV/HCV Protease Inhibitors against SARS-CoV-2 3CLpro. Antiviral Res. 2022, 207, 105419. [Google Scholar] [CrossRef]
- Sacco, M.D.; Hu, Y.; Gongora, M.V.; Meilleur, F.; Kemp, M.T.; Zhang, X.; Wang, J.; Chen, Y. The P132H Mutation in the Main Protease of Omicron SARS-CoV-2 Decreases Thermal Stability without Compromising Catalysis or Small-Molecule Drug Inhibition. Cell Res. 2022, 32, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.J.; Shi, Y. Effects of Data Quality and Quantity on Deep Learning for Protein-Ligand Binding Affinity Prediction. Bioorg. Med. Chem. 2022, 72, 117003. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
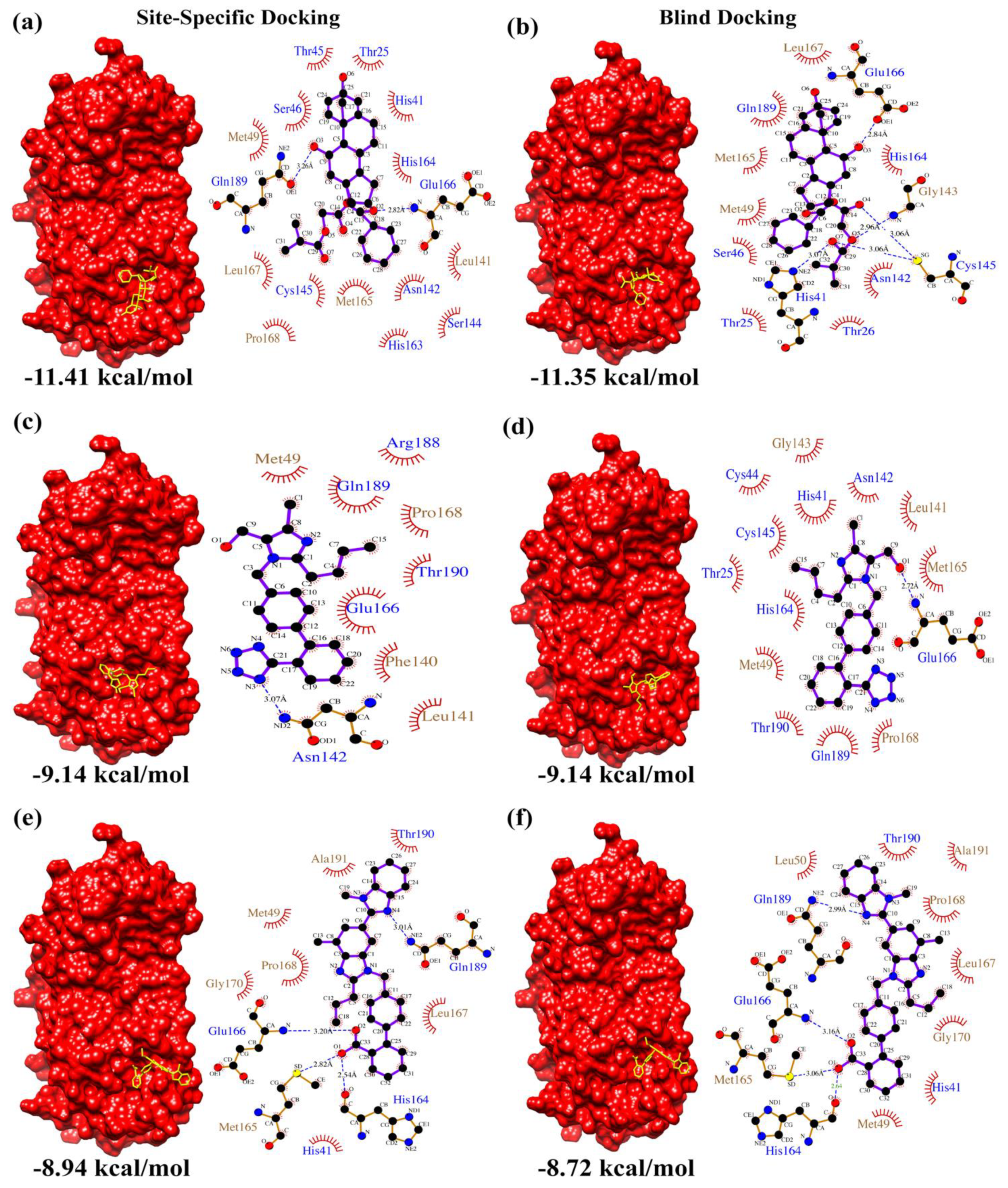
| e-Drug3D-lib | Ligand | Ciclesonide | Losartan | Telmisartan | |
| Site-Specific Docking | Calculated Binding Affinity Score (kcal/mol) | 1 | −11.41 | −9.14 | −8.94 |
| 2 | −11.06 | −9.14 | −8.29 | ||
| 3 | −11.03 | −9.14 | −8.02 | ||
| Blind Docking | Calculated Binding Affinity Score (kcal/mol) | 1 | −11.35 | −9.14 | −8.62 |
| 2 | −11.09 | −9.13 | −8.72 | ||
| 3 | −10.88 | −9.14 | −8.08 | ||
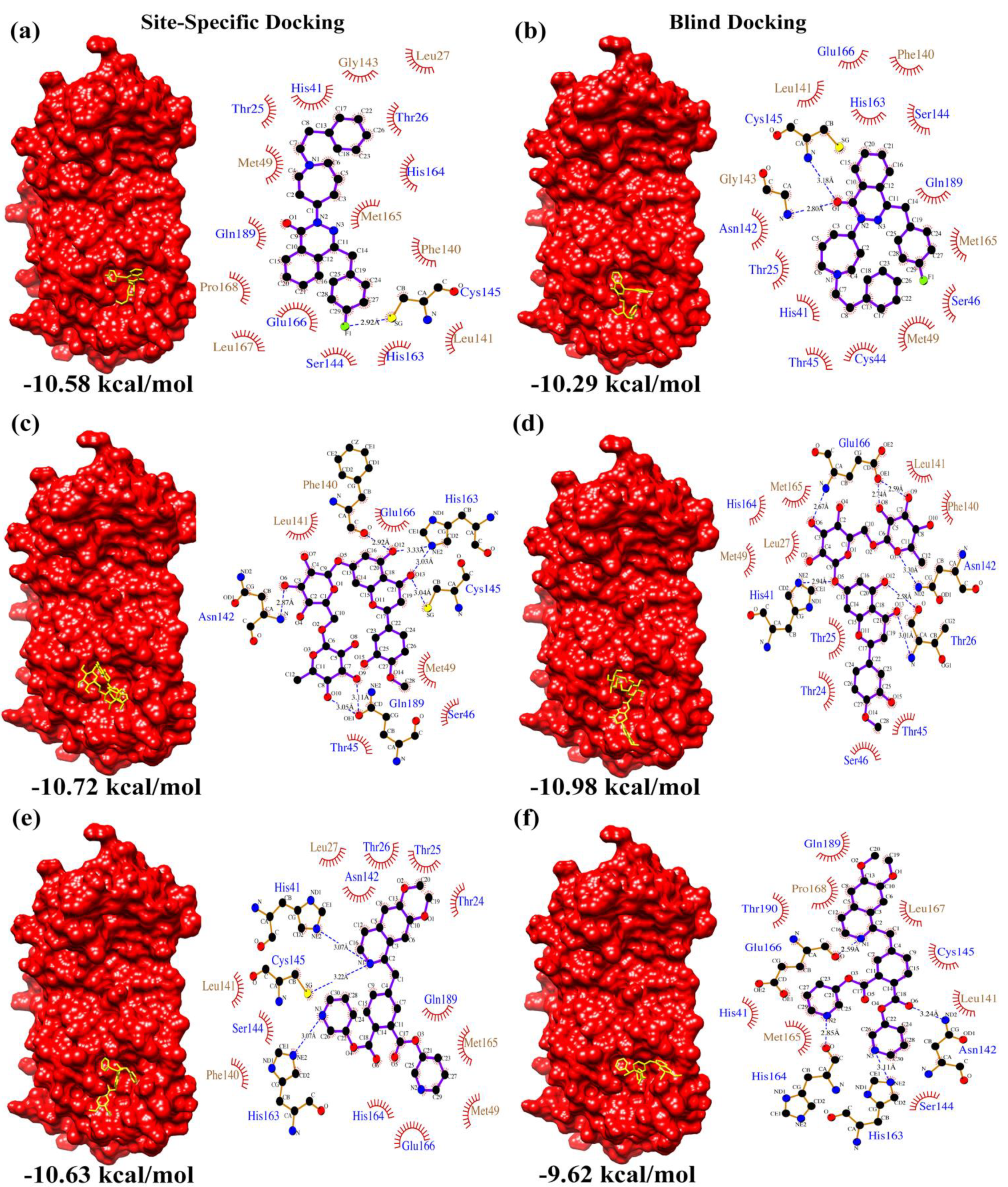
| Drugs-lib | Ligand | Flezelastine | Hesperidin | Niceverine | |
| Site-Specific Docking | Calculated Binding Affinity Score (kcal/mol) | 1 | −10.58 | −9.87 | −9.60 |
| 2 | −10.38 | −10.61 | −8.90 | ||
| 3 | −10.43 | −10.72 | −10.63 | ||
| Blind Docking | Calculated Binding Affinity Score (kcal/mol) | 1 | −10.29 | −10.98 | −9.62 |
| 2 | −10.10 | −9.78 | −9.05 | ||
| 3 | −10.00 | −9.16 | −9.42 | ||
| NP-lib | Ligand | NP1 | NP2 | NP3 | |
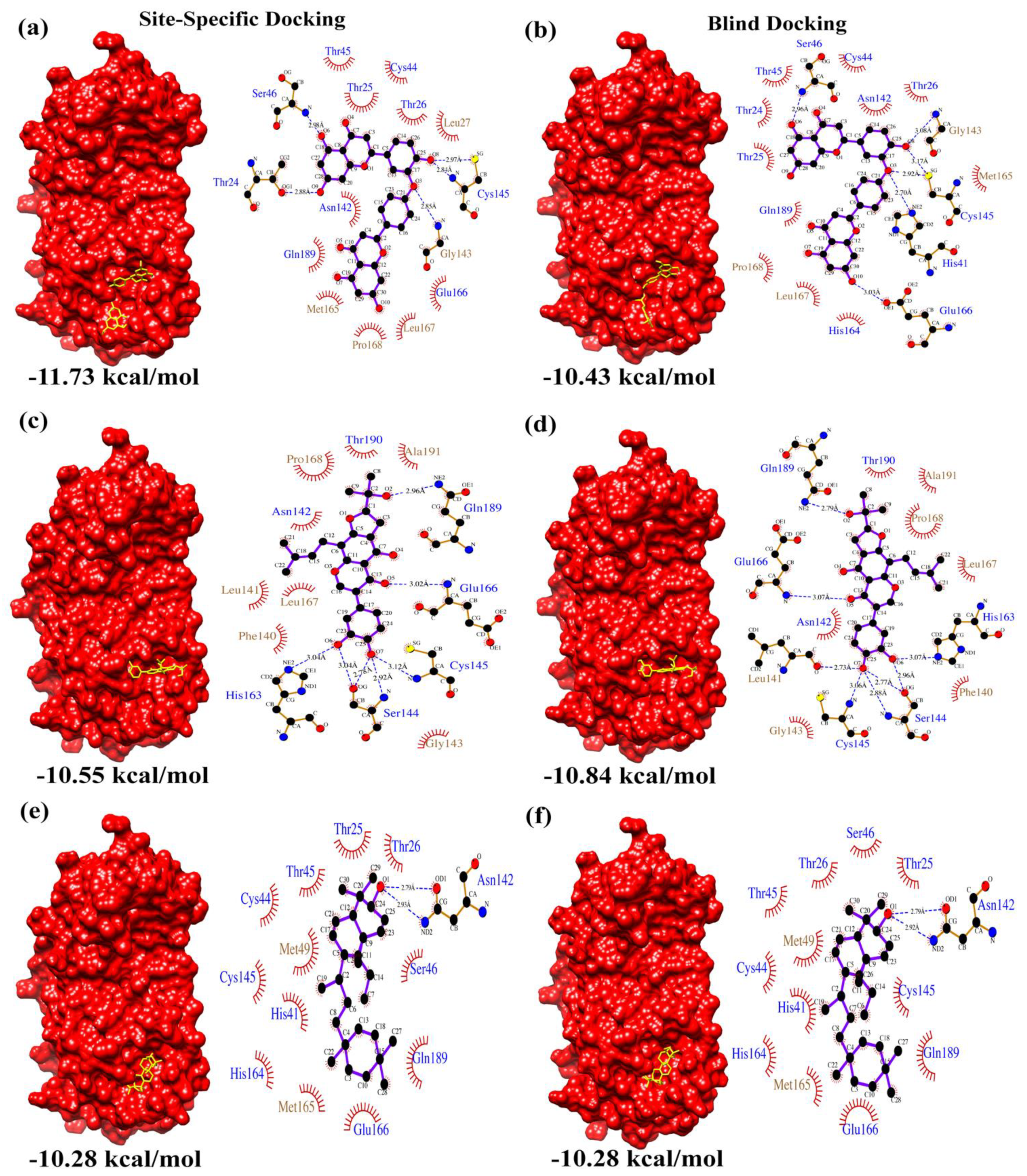
| Site-Specific Docking | Calculated Binding Affinity Score (kcal/mol) | 1 | −11.73 | −10.55 | −10.28 |
| 2 | −10.66 | −10.37 | −10.28 | ||
| 3 | −10.12 | −10.47 | −10.28 | ||
| Blind Docking | Calculated Binding Affinity Score (kcal/mol) | 1 | −10.43 | −10.70 | −10.28 |
| 2 | −10.14 | −10.84 | −10.28 | ||
| 3 | −10.34 | −10.65 | −10.28 | ||
| Drug Library | Drug/Ligand Name | Drug ID | Clinical Application/Use |
|---|---|---|---|
| e-Drug3D-lib | Ciclesonide | CAS 126544-47-6 | Anti-Asthma |
| Losartan | CAS 114798-26-4 | Anti-Hypertensive | |
| Telmisartan | CAS 144701-48-4 | Anti-Hypertensive | |
| Drugs-lib | Flezelastine | CAS 135381-77-0 | Anti-Asthma/Anti-Allergic |
| Hesperidin | CAS 520-26-3 | Antioxidant/Anti-Inflammatory | |
| Niceverine | CAS 2545-24-6 | Anti-Hypertensive | |
| NP-lib | NP1 (2,3,2″,3″-Tetrahydroochnaflavone) | CAS 678138-59-5 MolPort-039-052-621 | - |
| NP2 (Furowanin A) | CAS 911004-72-3 MolPort-039-141-993 | - | |
| NP3 (3S,6bS,8aR,12aR,12bS,14bS)4,4,6b,8a,11,11,12b,14b-octamethyl 1,2,3,4,4a,5,6,6b,7,8,8a,9,10,11,12,12a,12b,13,14,14b-icosahydropicen-3-ol | MolPort-002-527-314 | - |
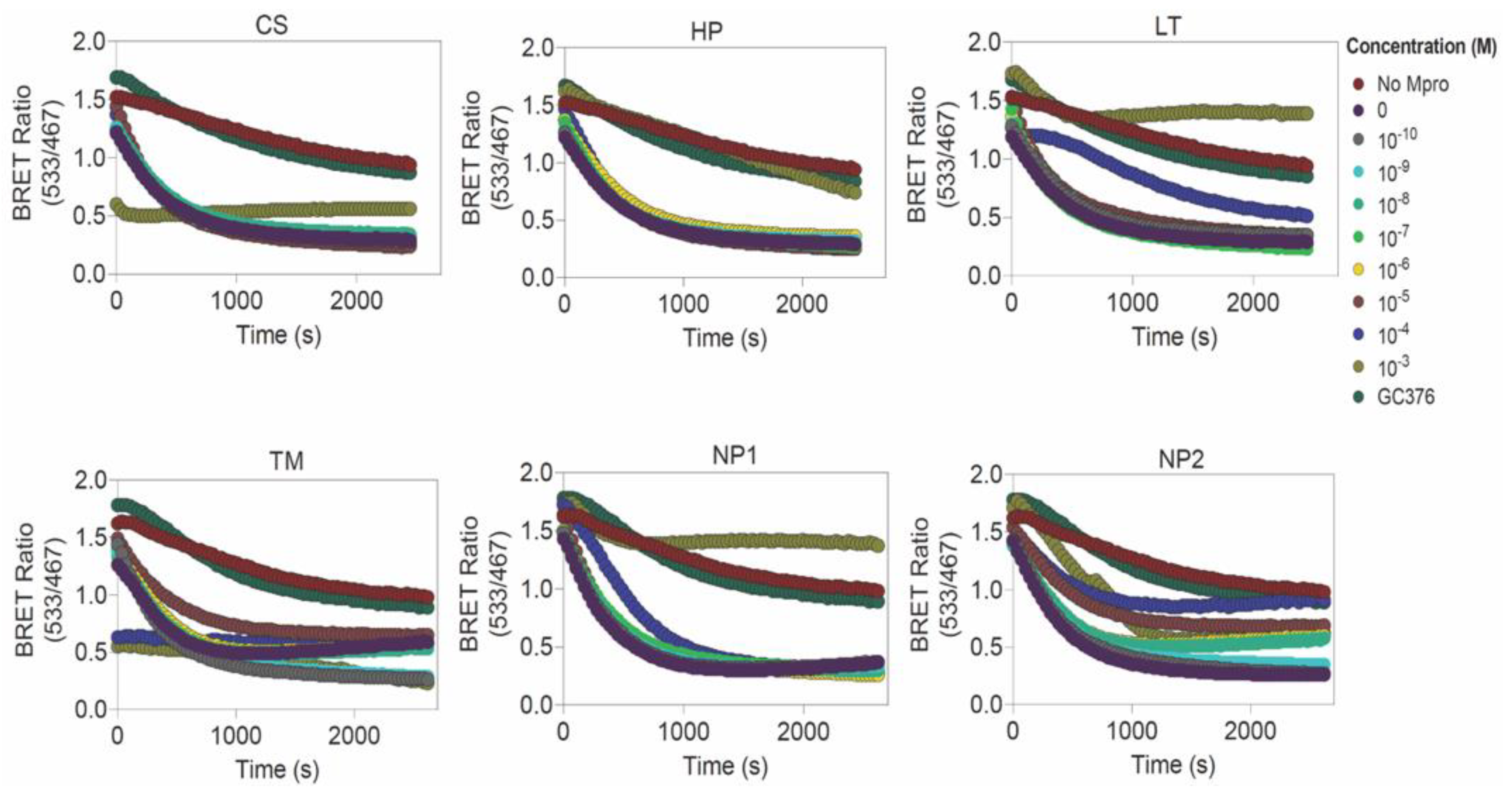
| Library | Drugs | Determined EC50 (μM) | Predicted IC50 (μM) | Blind Docking Score (kcal/mol) | Calculated Affinity (Kd) from Blind Docking Score (μM) | |
|---|---|---|---|---|---|---|
| e-Drug3D-lib | 1 | Ciclesonide (CS) | Not determined | 6.4 | −11.35 | 0.00469 |
| 2 | Losartan (LT) | 260.05 ± 88.60 | 9.11 | −9.14 | 0.196 | |
| 3 | Telmisartan (TM) | Not determined | 3.67 | −8.72 | 0.399 | |
| Drugs-lib | 1 | Flezelastine | Not tested | 2.04 | −10.29 | 0.0281 |
| 2 | Hesperidin (HP) | Not determined | 8.37 | −10.98 | 0.00876 | |
| 3 | Niceverine | Not tested | 3.4 | −9.62 | 0.0872 | |
| NP-lib | 1 | NP1 | 901.1 ± 10.60 | 23.19 | −10.43 | 0.0222 |
| 2 | NP2 | 124.8 ± 207.5 | 3.45 | −10.84 | 0.0111 | |
| 3 | NP3 | Not tested | 7.73 | −10.28 | 0.0286 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moovarkumudalvan, B.; Geethakumari, A.M.; Ramadoss, R.; Biswas, K.H.; Mifsud, B. Structure-Based Virtual Screening and Functional Validation of Potential Hit Molecules Targeting the SARS-CoV-2 Main Protease. Biomolecules 2022, 12, 1754. https://doi.org/10.3390/biom12121754
Moovarkumudalvan B, Geethakumari AM, Ramadoss R, Biswas KH, Mifsud B. Structure-Based Virtual Screening and Functional Validation of Potential Hit Molecules Targeting the SARS-CoV-2 Main Protease. Biomolecules. 2022; 12(12):1754. https://doi.org/10.3390/biom12121754
Chicago/Turabian StyleMoovarkumudalvan, Balasubramanian, Anupriya Madhukumar Geethakumari, Ramya Ramadoss, Kabir H. Biswas, and Borbala Mifsud. 2022. "Structure-Based Virtual Screening and Functional Validation of Potential Hit Molecules Targeting the SARS-CoV-2 Main Protease" Biomolecules 12, no. 12: 1754. https://doi.org/10.3390/biom12121754