Disrupting ACE2 Dimerization Mitigates the Infection by SARS-CoV-2 Pseudovirus
 Jiaqi Zhu
Jiaqi Zhu Yue Su
Yue Su Young Tang
Young Tang- Department of Animal Science, Institute of Systems Genetics, University of Connecticut, Storrs, CT, United States
The coronavirus disease 2019 pandemic has caused over million death and 500 million reported cases globally. More effective antiviral medications are needed to curb the continued spread of this disease. The infection by SARS-CoV-2 virus is initiated via the interaction between the receptor binding domain (RBD) of the viral glycoprotein Spike (S protein) and the N-term peptidase domain of the angiotensin-converting enzyme 2 (ACE2) expressed on the host cell membrane. ACE2 forms a protein homodimer primarily through its ferredoxin-like fold domain (a.k.a., Neck-domain). We investigated whether the dimerization of ACE2 receptor plays a role in SARS-CoV-2 virus infection. We report here that the ACE2 receptor dimerization enhances the recognition of SARS-CoV-2 S protein. A 43-amino-acid peptide based on the N-term of Neck-domain could block the ACE2 dimerization and hence the interaction between RBD and ACE2 and mitigate the SARS-CoV-2 S protein pseudotyped virus/host cell interaction. Our study illustrated a new route to develop potential therapeutics for the prevention and treatment of SARS-CoV-2 viral infection.
Introduction
The COVID-19 pandemic causes severe respiratory syndrome and mortality especially in adult and elderly human beings and has paralyzed the global societal and economic development and interactions. The entry of SARS-CoV-2 virus in host cells starts with the recognition of its S protein by the angiotensin-converting enzyme 2 (ACE2) on the cell membrane, which triggers the cleavage of Spike (S protein) into two subunits (S1 and S2, Figure 1) by host cell proteases (1, 2), with a subsequent membrane fusion between virus and host cells driven by the S2 subunit. In addition to the development of vaccines, many efforts have focused on blocking the viral entry by targeting the receptor binding domain (RBD) of S protein or the N-term peptidase domain (PD) of ACE2 with neutralizing antibodies, recombinant or soluble ACE2, peptides based on the PD of ACE2 and RBD region of S protein, and small molecules targeting these domains (3–5), with additional focus on the development of peptides and protease inhibitors blocking viral fusion and replication and host cell endosomal and some other pathways (3, 4).
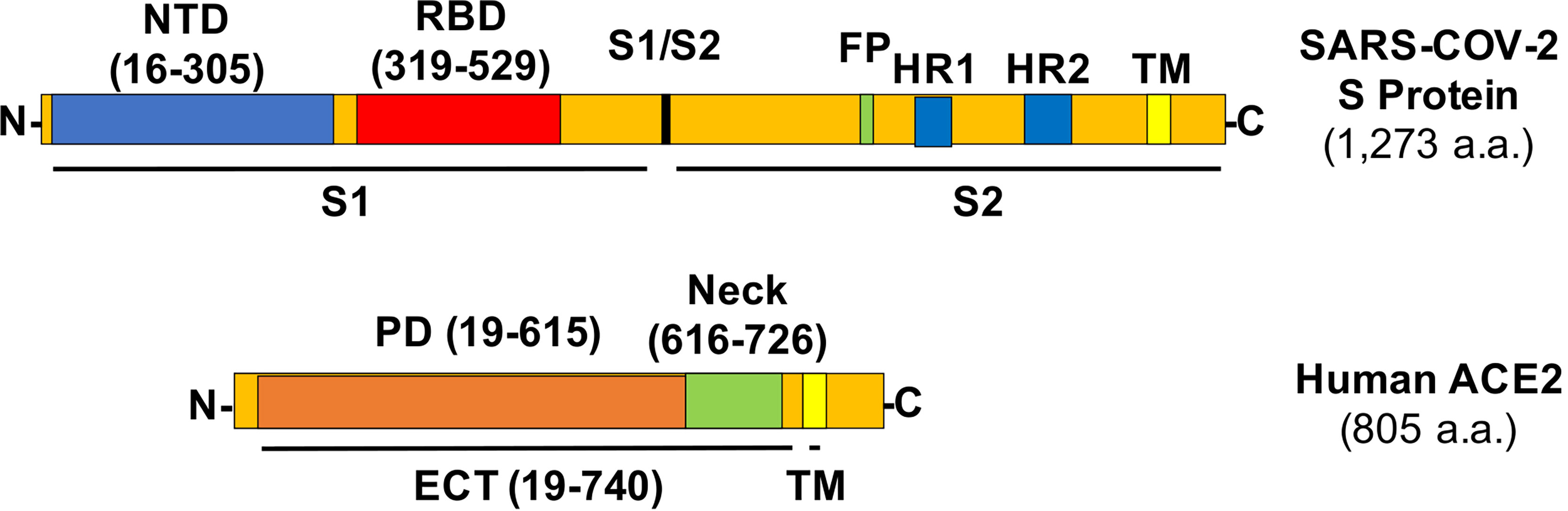
Figure 1 Structures of the SARS-CoV-2 S protein and human ACE2 with relevant amino acid residues. NTD, N-term domain; FP, fusion peptide; HR, heptad repeat; TM, transmembrane helix.
The S protein in the SARS-CoV-2 viral envelop forms a homotrimer with the rotation of one of the three RBDs from “down” to “up” position to be accessible by the PD of ACE2 (6, 7). The ectodomain (ECT) region of ACE2 consists of PD and the ferredoxin-like fold domain (a.k.a., Neck-domain) (8). The crystal structure analysis revealed that the ACE2 receptor naturally forms a homodimer between their Neck-domains, with an additional “closed” or “open” conformation developed via either the formation or the break of another weak interaction in the PD region of the protein dimer (8). However, the significance of this dimerization for the ACE2/S protein interaction is unclear. In this study, we found that ACE2 dimerization plays a previously unrecognized role in ACE2/S protein interaction, and inhibiting this dimerization-mitigated viral infection. Our study will have an important impact on the development of new therapeutics to modify the sensibility of host cells to SARS-CoV-2 virus infection.
Materials and Methods
DNA Constructs, Cell Culture, and Virus Production
For BiFC assay, the RBD domain in S protein of SARS-CoV-2 virus was inserted into pBiFC-VN155 (I152L) vector; ACE2 and its ECT/PD domains were cloned into pBiFC-VN155 (I152L) and pBiFC-VC155 vectors (9) (Addgene, MA, USA) (see Table 1 for the cloning primer details). The RBD, Neck-domain, and different Neck-domain fragments followed directly with a stop codon were cloned into the pBiFC-VN155 (I152L) vector. For protein expression, the cell membrane-penetrating peptide (TAT), red fluorescence protein (DsRed), and NK-NT or NKN1 fragments were cloned into pET6xHN-N Vector (Takara, CA, USA) (see Table 2 for the cloning primer details). HEK293T cells were cultured in FP medium (Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 2 mM GlutaMAX™ supplement, 0.1 mM MEM non-essential amino acids, and 50 U/ml and 50 μg/ml penicillin–streptomycin, respectively). For ACE2-expressing lentivirus packaging, pscALPSpuro-HsACE2 (human) (Addgene, MA, USA) was co-transfected with psPAX2 and pCMV-VSV-G packaging plasmids into HEK293T cells using FuGENE 6 (Promega, WI, USA). The supernatants containing the virus were collected at 48 and 72 h after transfection. For doxycycline (Dox) inducible, S protein-pseudotyped luciferase-expressing lentivirus preparation, HEK293T cells were transfected with FUW-RLuc-T2A-PuroR (10) (Addgene, MA, USA), psPAX2, and pUNO1-SARS2-S (D614G) (Invivogen CA) packaging plasmids using FuGENE 6. The supernatants containing the virus were collected at 48 and 72 h after transfection. To establish ACE2-expressed cell line (ACE2-HEK293T cells), HEK293T cells were infected with ACE2-expressing lentivirus, and ACE2-positive cells were selected by 2 ug/ml of puromycin.
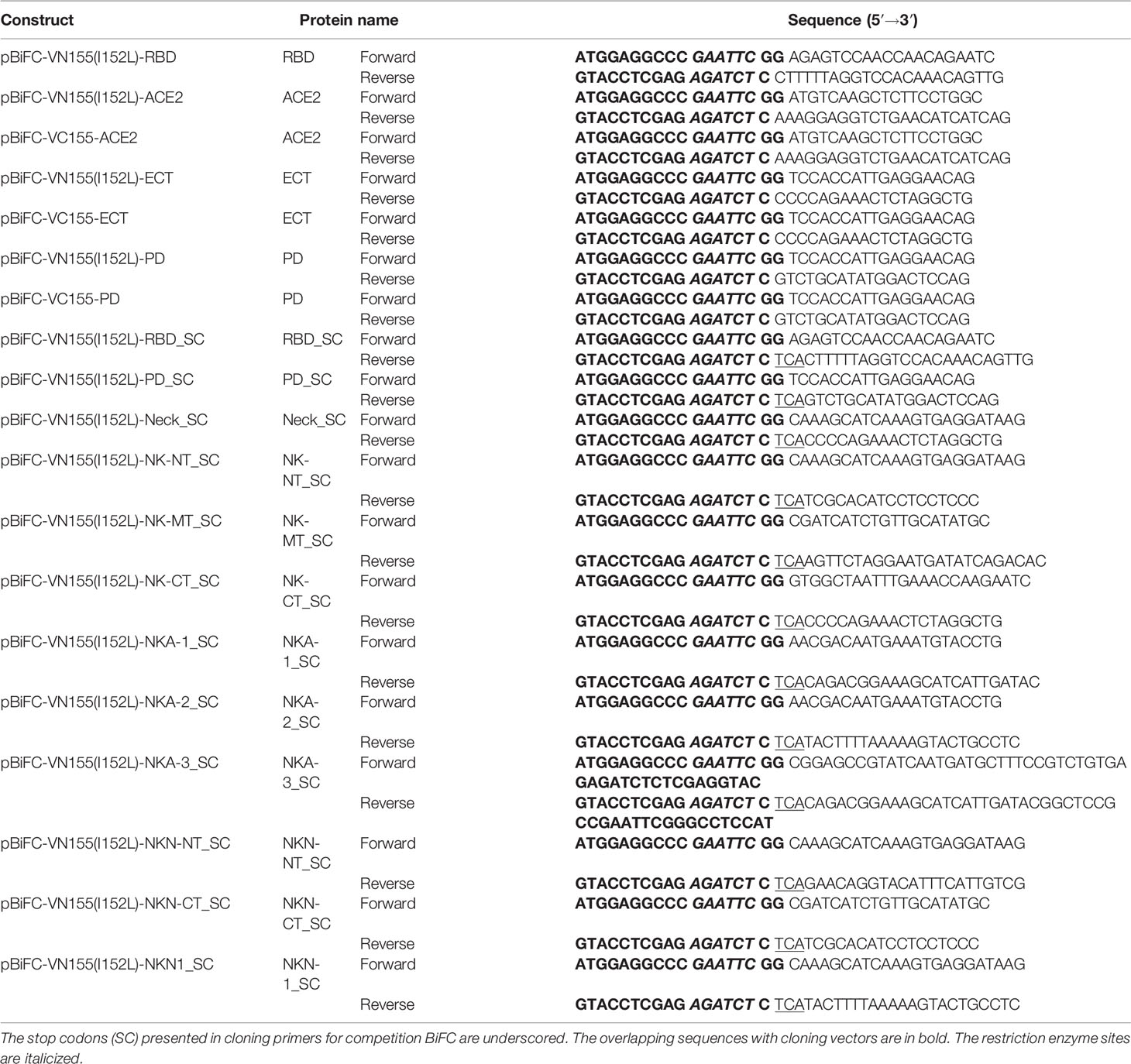
Table 1 Cloning primers for the BiFC assays.

Table 2 Cloning primers for recombinant protein expression.
Bimolecular Fluorescence Complementation (BiFC) Assay
For the bimolecular fluorescence complementation (BiFC) assay, the HEK293T cells were cultured in 12-well plates and were co-transfected with 0.5 μg of each construct expressed in pBiFC-VN155 (I152L) and pBiFC-VC155 vectors per well using FuGENE 6. In competition BiFC assay, HEK293T cells were co-transfected with 0.5 μg of each construct expressed in pBiFC-VN155 (I152L) and pBiFC-VC155 vectors, together with 5 μg competitor constructs with stop codon in pBiFC-VN155 (I152L) vector. For the protein competition BiFC assay, HEK293T cells were co-transfected with 0.5 μg of each construct expressed in pBiFC-VN155 (I152L) and pBiFC-VC155 vectors in 12-well plates. FP medium containing 0.1, 1, or 2 μg of recombinant TAT-Dsred-GFP/NK-NT/NKN1 proteins was added to the cells at 4 h after transfection. Fluorescence images were taken at 24 and 48 h after transfection using a Nikon fluorescence microscope, and the fluorescence intensity was quantified by Image J.
Protein Expression and Purification
The recombinant pET6xHN-N constructs were transformed into Escherichia coli strain BL21 (DE3) (New England Biolabs, MA, USA). The transformed clones were cultured at 37°C in LB broth with 100 μg/ml carbenicillin, induced by adding 1 mM isopropyl β-D-1-thiogalactopyranoside at optical density of 0.6–0.8, and then incubated at 23°C overnight. For protein purification, cells were harvested by centrifugation at 5,000 rpm for 15 min and resuspended in xTractor Buffer containing DNAse I, lysozyme solution, and Protease Inhibitor Cocktail (Takara Bio USA, Inc., CA). The suspension was sonicated for 10 s three times with a 30s pause and centrifuged at 10,000 rpm for 20 min. The supernatant was incubated with equilibrated TALON Metal Affinity Resin (Takara Bio USA, Inc., CA, USA) for 20 min on ice. The proteins were eluted from resin with the elution buffer (pH 7.0, 150 mM imidazole, 50 mM NaH2PO4, and 300 mM NaCl). The eluted proteins were concentrated and with buffer exchanged to phosphate-buffered saline (pH 7.4) using Protein Concentrator PES, 3K MWCO (Pierce Biotechnology, PA, USA).
Co-Immunoprecipitation
For HEK293T, cells in a 6-well-plate were transfected with 1 μg Myc-tagged ECT-expressing plasmids per well, and the total proteins were extracted from the cells by adding Cell Lysis Buffer (Cell Signaling Technology, MA, USA) containing 500 mM NaCl, 1.5% Triton X-100 and 1 mM PMFS at 48 h after transfection. Supernatant was collected after centrifugation at 14,000 × g for 10 min and was incubated at 4°C overnight with 5 μg of recombinant DsRed, NK-NT-DsRed, or NKN1-DsRed proteins and Myc-Tag (9B11) Mouse mAb (1:1,000, Cell Signaling Technology, MA, USA). Moreover, 30 μl of protein G Agarose Beads (Cell Signaling Technology, MA, USA) slurry was added to the protein mixture, and the latter was rotated at 4°C for 3 h. The agarose beads were washed with cell lysis buffer 5 times and then resuspended in 30 μl of NuPAGE™ LDS Sample Buffer (4X) and 12 μl NuPAGE™ Sample Reducing Agent (10X) (Invitrogen, MA, USA).
Western Blotting
The samples collected from immunoprecipitation were separated by 10% SDS-PAGE gel and transferred onto an Immobilon-P polyvinylidene fluoride (PVDF) membrane. The membrane was blocked by 5% skimmed milk and incubated at 4°C overnight with ACE-2 Antibody (1:2,000, Novus Biologicals, CO, USA) and 6xHN Polyclonal Antibody (1:2,000, Takara, CA, USA). The membrane was then incubated with secondary horseradish peroxidase (HRP)-linked, anti-rabbit IgG (1:10,000, Cell Signaling Technology, MA, USA) for 1.5 h at room temperature.
Proteins extracted from HEK293T cells in the BiFC assay were separated by 10% SDS-PAGE gel and transferred onto an Immobilon-P PVDF Membrane. The membrane was incubated at 4°C overnight with ACE-2 Antibody (1:2,000, Novus Biologicals, CO, USA), Myc-Tag (9B11) Mouse mAb (1:1,000, Cell Signaling Technology, MA), and GAPDH (D16H11) XP® Rabbit mAb (1:1,000, Cell Signaling Technology, MA, USA). The membrane was then incubated with secondary HRP-linked, anti-rabbit IgG (1:10,000, Cell Signaling Technology, MA, USA), and goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody, HRP (1:2,000, Thermo Fisher Scientific, MA, USA) for 1.5 h at room temperature. Images were developed with Clarity™ Western ECL Substrate (Bio-Rad Laboratories, CA, USA) and visualized under the ChemiDox XRS Image System (Bio-Rad Laboratories, CA, USA).
mRNA Transfection and S Protein Pseudovirus Luciferase Assay
For mRNA preparation, GFP, NK-NT, and NTN1 DNA fragments harboring T7 promoter sequence were amplified by PCR, purified, and transcribed to mRNAs in vitro using mMESSAGE mMACHINE™ T7 ULTRA Transcription Kit (Invitrogen, CA, USA) (see Table 3 for PCR primer details). For the S protein pseudovirus infection and luciferase assay, ACE2-expressing HEK293T cells were seeded into 24-well plates and transfected with 2 μg of each mRNA/well using Lipofectamine™ MessengerMAX™ Transfection Reagent (Invitrogen, CA, USA). After 24 h, the cells were infected with Spike-RLuc pseudotyped lentivirus, M2rtTA lentivirus, and 10 ug/ml Polybrene for 1 h. The luciferase expression was induced by adding 1 μg/ml doxycycline at 24 h after viral infection. Luciferase activity in cells was measured using Renilla Luciferase Glow Assay Kit (Pierce Biotechnology, PA, USA) and the CLARIOstar Plus plate reader (BMG LABTECH, NC, USA) at 24 h later.

Table 3 PCR primers for mRNA templates.
Statistial Analysis
One-way ANOVA with Tukey’s multiple-comparison post-hoc test or Student’s t-test was used for data analysis. The figures were presented as mean ± standard deviation. A p-value <0.05 was considered statistically significant.
Results
Neck-Domain Is Needed for ACE2 Receptor Dimerization in Living Cells
The ACE2 ECT region consists of PD, the Neck-domain, and a long linker before the transmembrane helix (TM, excluded from ECT) (Figure 1). The intensive polar interaction between Neck-domains is believed to primarily mediate ACE2 dimerization based on cryo-EM structural analysis (8). The bimolecular fluorescence complementation (BiFC) assay based on the reconstitution of the N- and C-term fragments of the YFP protein Venus (VN and VC) has been widely applied for protein–protein interaction (PPI) study under physiological conditions (9, 11), allowing a direct visualization of PPI in living cells with high sensitivity and without exogenous reagent and complicated data process (12, 13). To verify the function of Neck-domain in living cells, we designed BiFC assays to evaluate the Neck-domain-mediated ACE2 dimerization (Figures 2A, B). ACE2 PD (without Neck-domain) fused with VN or VC were co-expressed in HEK293T cells. Alternatively, ACE2 ECT (with Neck-domain) fused with VN or VC were co-expressed. As expected, the co-expression of PD-VN and PD-VC yielded no fluorescence, while the co-expression of ECT-VN and ECT-VC produced a strong fluorescence (Figures 2C, D). The co-expression of ECT-VN with PD-VC or ECT-VC with PD-VN did not also yield any significant fluorescence signal (Figures 2C, D). These data in living cells support the notion that Neck-domain is critical for ACE2 protein dimerization (8).
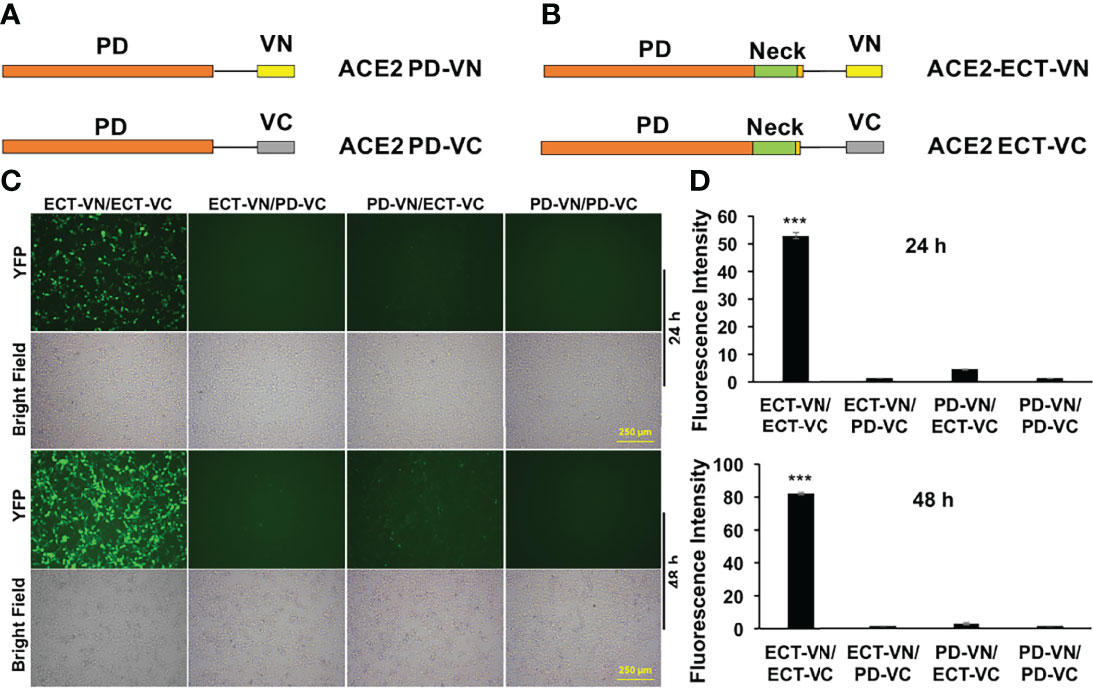
Figure 2 BiFC assays to verify the Neck-domain function on ACE2 dimerization. (A) The PD is fused with the N-term or C-term fragment of YFP protein Venus (VN or VC). (B) The ECT is fused with VN or VC. (C) Representative fluorescence images of HEK293T cells co-expressing different combinations of the ACE2-ECT and ACE2-PD constructs at 24 and 48 h after the plasmid transfection. Bar = 250 µm. (D) Quantitation of the fluorescence intensity at 24 h (upper) and 48 h (lower) after plasmid transfection. p < 0.001, N = 3. ***p < 0.001.
Neck-Domain Is Needed for the ACE2/S Protein Interaction
To investigate the significance of Neck-domain for the PPI between human ACE2 and SARS-CoV-2 S protein, we developed BiFC assay constructs by fusing S protein RBD with VN and ACE2 PD or ECT with VC (Figure 3A). Western blotting (WB) confirmed the co-expression of S protein RBD-VN and ACE2 PD-VC or ECT-VC fusion proteins in HEK293T cells (Figure 3B). The YFP fluorescence was then quantified at 24 h after plasmid transfection. Intriguingly, the BiFC assay revealed that ACE2 PD (without Neck-domain) was incapable of binding to S protein RBD in living cells, with the background fluorescence signal similar to a pair of previously reported non-interactive proteins (14)—CD163 scavenger receptor cysteine-rich domain 2 (SRCR2) and porcine reproductive and respiratory virus glycoprotein GP2a (Figure 4). On the contrary, ACE2 ECT which contains Neck-domain showed a strong interaction with S protein RBD (Figure 4). These results strongly indicate that the presence of Neck-domain is necessary for the recognition of viral S protein by ACE2. Based on the fact that Neck-domain is critical for ACE2 dimerization, our results here support the hypothesis that Neck-domain is needed for the ACE2 recognition of S protein, most likely through mediating the dimerization of ACE2.

Figure 3 Design of the BiFC assay to detect the ACE2/S protein PPI. (A) The RBD of S protein was fused with VN, and PD (a.a. 19–615) or ECT (a.a. 19–740) of ACE2 was fused with VC. (B) Western blot detection of the recombinant proteins co-expressed in HEK293T cells for BiFC assay using specific antibodies. GAPDH served as the loading control.
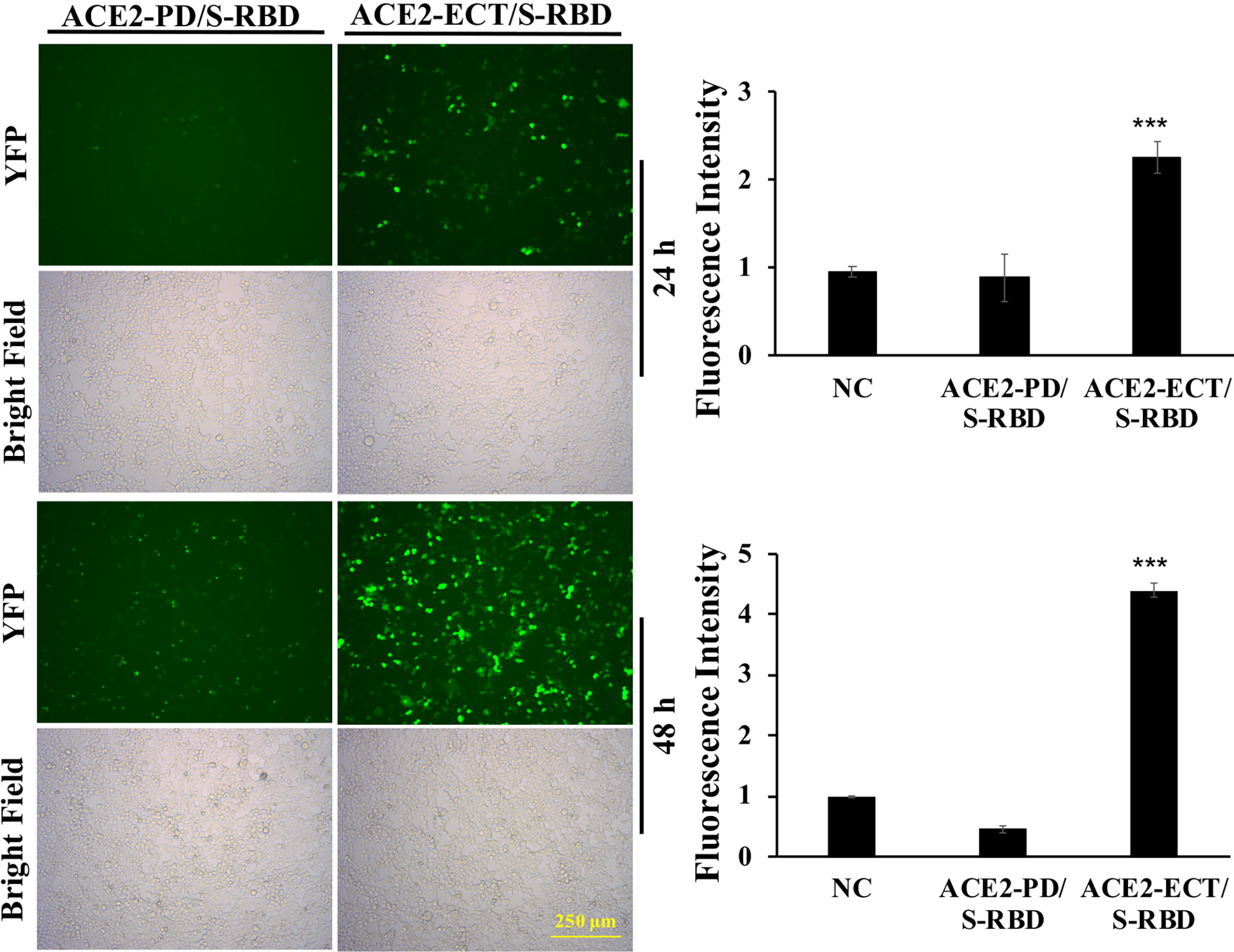
Figure 4 ACE2 Neck-domain is critical for ACE2/S protein interaction. Left: representative BiFC fluorescence images of HEK293T cells co-transfected with ACE2-PD-VC or ACE2-ECT-VC and S protein RBD-VN plasmids at 24 and 48 h, respectively. Bar = 250 µm. Right: quantitation of the relative fluorescence intensity. p < 0.001, N = 3. NC, negative control using a pair of non-interactive proteins (CD163-SRCR2 and GP2a) as described in the main text. ***p < 0.001.
Screening Parts of Neck-Domain Critical for ACE2 Dimerization and S Protein Recognition
To investigate which part of Neck-domain is critical for ACE2 dimerization, we designed a competition assay by co-expressing different segments of Neck-domain as the competitor (Figure 5A) in the ECT-VN/ECT-VC BiFC system (Figure 2B). The competitor plasmids were added at 5-fold of ECT-VN/ECT-VC plasmids, with data collected at 72 h after plasmid transfection to allow the proper expression of ECT-VN/ECT-VC plasmids. As expected, PD without Neck-domain failed to interfere with the interaction between ECT-VN and ECT-VC (Figure 5B). The competition BiFC screening revealed that N-term residues 616–671 of Neck-domain (NK-NT, Figure 5A) strongly inhibited the PPI between ECT-VN and ECT-VC (Figure 5B). Interestingly, the overexpression of NK-MT caused the aggregation and death of cells (Figure 5B). To investigate how the disturbance of ECT dimerization could impact the PPI between S protein RBD and ECT, we co-expressed these segments of Neck-domain as competitors in the RBD-VN/ECT-VC BiFC system (Figure 3A). Over-expression of PD as a competitor could only slightly inhibit the recognition of RBD by ECT (Figure 5C). However, the NK-NT fragment exhibited the most significant inhibitory effect on the PPI between RBD and ECT (Figures 5C, D), similar to the ECT dimerization BiFC assay (Figure 5B). Further screening of different parts of NK-NT identified that residues 616–658 of Neck-domain (NKN1, Figure 5A) shared a similar potency as NK-NT in blocking the RBD/ECT interaction (Figure 5E). This correlates well with the comparable efficiency on inhibition of full-length ACE2 protein dimerization by NK-NT and NKN1 in BiFC assay (Figure 5F).
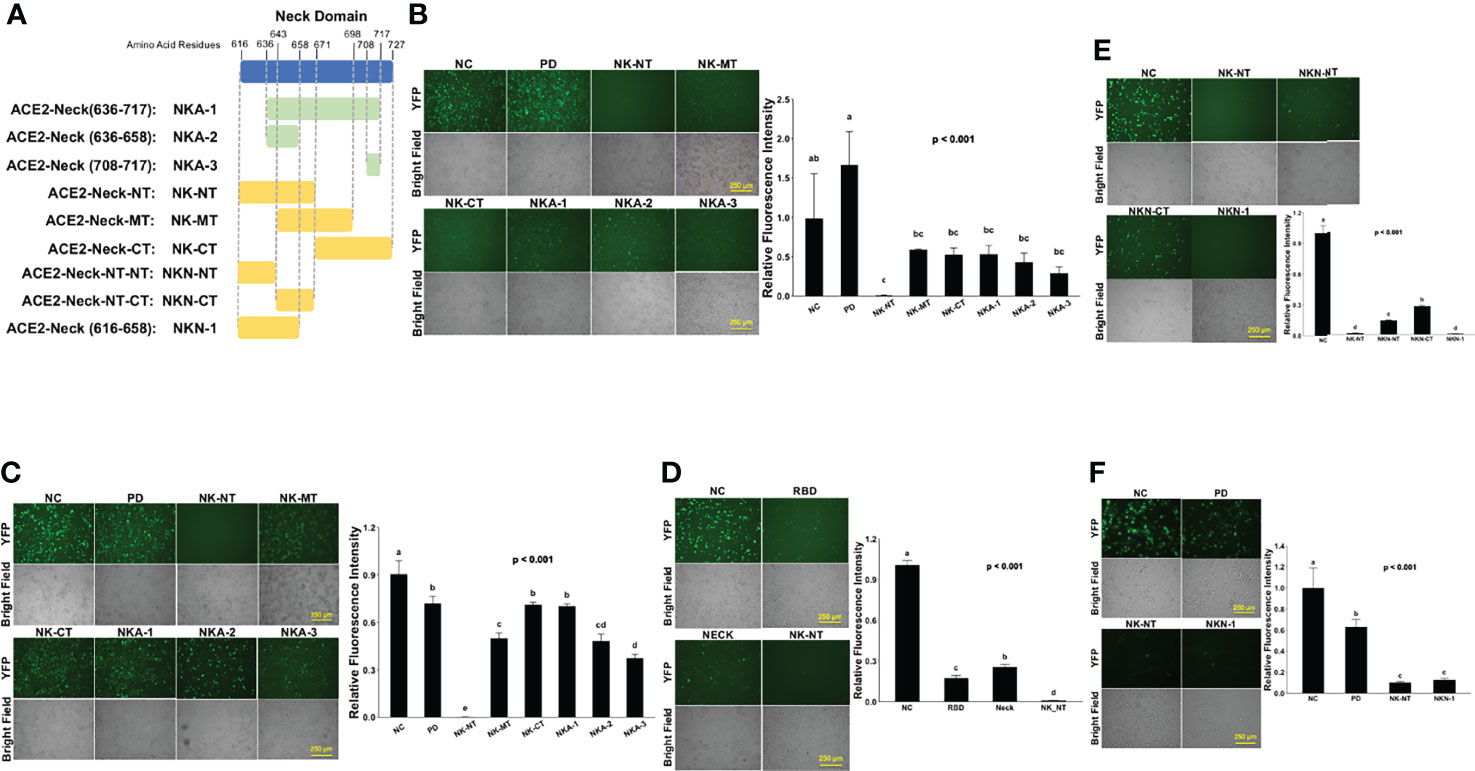
Figure 5 Screening parts of Neck-domain critical for ACE2 dimerization and recognition of S protein. (A) Schematic diagram of different Neck-domain fragments for BiFC screening. (B) Competition ECT-VN/ECT-VC BiFC results with the co-expression of different Neck-domain fragments as competitors. Left: representative BiFC fluorescence images. Bar = 250 µm. Right: quantitation of the relative fluorescence intensity. p < 0.01, N = 3. NC, negative control of ECT-VN/ECT-VC BiFC with no competitors. (C) Competition RBD-VN/ECT-VC BiFC results with the co-expression of different neck domain fragments as competitors. Left: representative BiFC fluorescence images. Bar = 250 µm. Right: quantitation of the relative fluorescence intensity. p < 0.001, N = 3. NC, negative control of RBD-VN/ECT-VC BiFC with no competitors. (D) Competition RBD-VN/ECT-VC BiFC results with RBD, Neck-domain, and NK-NT as competitors. Left: representative BiFC fluorescence images. Bar = 250 µm. Right: quantitation of the relative fluorescence intensity. p < 0.001, N = 3. (E) Comparing competition RBD-VN/ECT-VC BiFC results with additional fragments of NK-NT as competitors. Left: representative BiFC fluorescence images. Bar = 250 µm. Right: quantitation of the relative fluorescence intensity. p < 0.001, N = 3. (F) Comparing competition full-length ACE2-VN/ACE2-VC BiFC results with PD, NK-NT, and NKN1 as competitors. Left: representative BiFC fluorescence images. Bar = 250 µm. Right: quantitation of the relative fluorescence intensity. p < 0.001, N = 3. Different letters indicate significant difference between conditions.
In order to double-verify the results of the competition BiFC assays, we engineered histidine and asparagine (HN)-tagged protein constructs with fusion of a cell-penetrating peptide TAT (15), red fluorescence protein DsRed, and NK-NT (Figure 6A, upper). The TAT-DsRed (16) and TAT-DsRed-NK-NT recombinant proteins were purified, quantified, and added directly to a culture medium of 293T cells transfected with ECT-VN/ECT-VC or ACE2-VN/ACE2-VC plasmids (Figure 6A, lower). Compared with the TAT-DsRed control, 1 and 2 μg/ml TAT-DsRed-NK-NT significantly, though not completely, inhibited the dimerization of ECT (Figure 6B) or ACE2 (Figures 6C, D). We also expressed HN-tagged DsRed-NKN1 and DsRed-NK-NT fusion proteins (Figure 6E) and conducted a co-immunoprecipitation (Co-IP) experiment using the total lysates of 293T cells over-expressing Myc-tagged ECT. We found that both NKN1 and NK-NT recombinant proteins interacted with ECT (Figure 6F). Taken together, our results demonstrated a 43-a.a. peptide (NKN1) as the critical segment in Neck-domain necessary for the PPI between ACE2/S protein by mediating ACE2 dimerization.
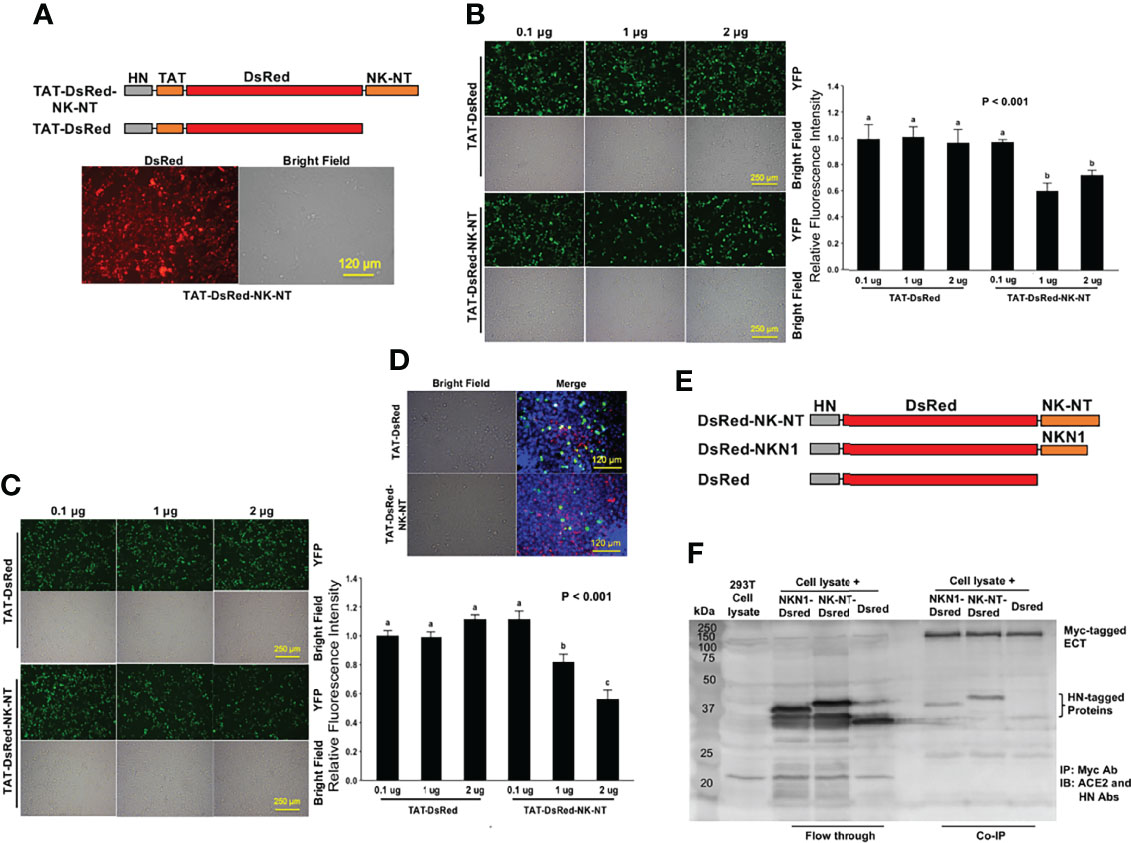
Figure 6 Recombinant NK-NT and NKN1 proteins bind to ECT and block ACE2 dimerization. (A) Upper: schematic diagram of histidine and asparagine (HN)-tagged recombinant protein constructs with cell-penetrating peptide TAT. Lower: representative image showing the HN-TAT-NK-NT recombinant protein transfected into HEK293T cells. Bar = 120 µm. (B) Competition ECT-VN/ECT-VC BiFC results with the TAT-DsRed control or TAT-DsRed-NK-NT as competitors. Left: representative BiFC assay fluorescence images. Bar = 250 µm. Right: quantitation of the relative fluorescence intensity. p < 0.001, N = 3. (C) Competition ACE2-VN/ACE2-VC BiFC assay results with the TAT-DsRed control or TAT-DsRed-NK-NT as competitors. Left: representative BiFC fluorescence images. Bar = 250 µm. Right: quantitation of the relative fluorescence intensity. p < 0.001, N = 3. (D) Overlay of fluorescence images in ACE2-VN/ACE2-VC BiFC assay with TAT-DsRed or TAT-DsRed-NK-NT as competitors. Green, Venus; red, DsRed recombinant proteins; blue, DAPI nuclear stain. Bar = 120 µm. (E) Schematic diagram of HN-tagged recombinant NK-NT and NKN1 protein constructs. (F) Co-IP results of purified HN-tagged recombinant DsRed, NK-NT, and NKN1 proteins incubated with the lysates of HEK293T cells expressing Myc-tagged ECT. Different letters in (B ,C) indicate significant difference among conditions.
NK-NT and NKN1 mRNAs Inhibit SARS-CoV-2 Viral Infection
A previous cryl-EM structure study mapped extensive polar interactions for ACE2 dimerization in the second (a.a. 636–658, NKA-2) and fourth (a.a. 708–717, NKA-3) helices of Neck-domain (8) (Figure 7A), which may explain the partial activities that we observed for NKA-2 and NKA-3 segments in blocking ECT dimerization (Figure 5B). However, NK-NT and NKN1 fragments exhibited the greatest potency in blocking ECT dimerization (Figure 5B), indicating that, in addition to the second helix, the additional residue(s) before that (residues 616–636) also has a critical function for ACE2 dimerization (Figure 7A). One such could be Y633 that forms a cation-π interaction with R710 in the fourth helix (8) (Figure 7A). In order to test whether the expression of NK-NT and NKN1 fragments can interfere with the SARS-CoV-2 viral infection, we in vitro transcribed and purified the NK-NT and NKN1 mRNAs, with GFP mRNA as the negative control (Figure 7B). In total, 2 μg mRNAs per well were transfected to 293T cells stably expressing human ACE2 in 24-well plates. After 24 h of transfection, the cells were infected with Renilla luciferase-expressing FUW-lentivirus pseudotyped with SARS-CoV-2 S protein (D614G) as the envelope protein, and luciferase intensity was evaluated at 48 h after viral infection. We found that compared with the GFP mRNA, transfection of both NK-NT and NKN1 mRNAs significantly inhibited the S protein pseudotyped lentivirus infection of the host cells (Figure 7C).
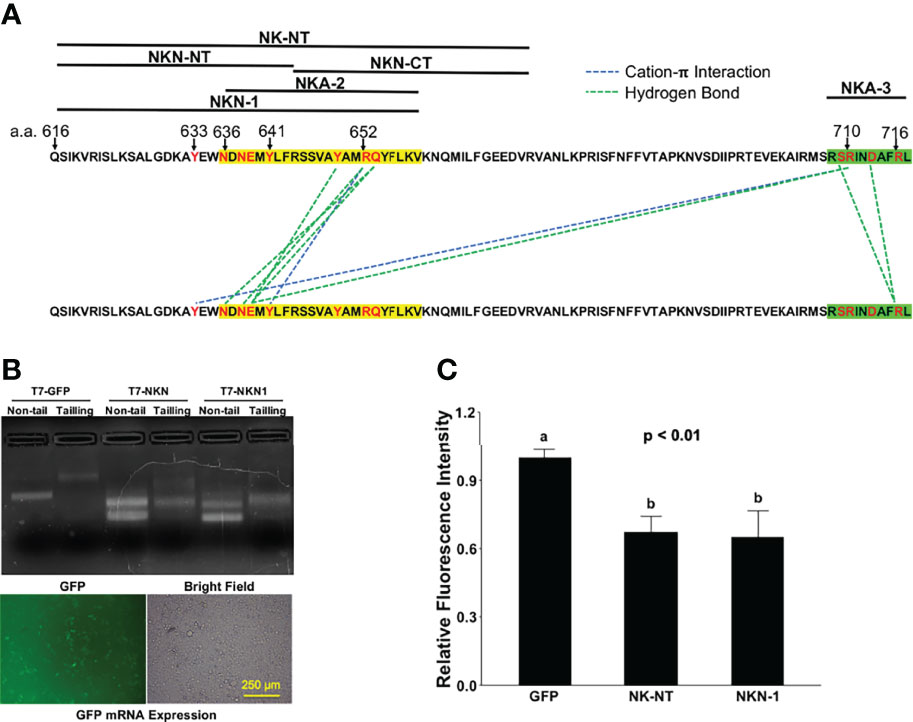
Figure 7 NK-NT and NKN1 mRNAs mitigate SARS-CoV-2 pseudovirus infection. (A) Residues in NK-NT, NKN1, and Neck-domain critical for ACE2 dimerization. The second and fourth helices are highlighted by yellow and green, respectively. (B) Upper: GFP, NK-NT, and NKN1 mRNAs transcribed in vitro via DNA constructs with a T7 promoter. Non-tail, purified mRNAs transcribed in vitro; tailing, purified mRNAs with the addition of poly-A tail; lower, expression of GFP mRNA after transfection into HEK293T cells. Bar = 250 µm. (C) Relative luminescence intensity in HEK293T cells in 24-well plates transfected with 2 µg of GFP, NK-NT, or NKN1 mRNAs per well and followed by infection of S protein pseudotyped lentivirus expressing Renilla luciferase. p < 0.01, N = 3. Different letters indicate significant difference between conditions.
Discussion
The COVID-19 pandemic has caused over 500 million infection cases and 6 million deaths globally. Numerous research have been done to better understand the mechanism of SARS-CoV-2 viral infection and its pathological effect in order to combat this disease. In this study, we tried to understand the fundamental aspects that determine this virus/host cell interaction to aid in the design of effective therapeutics, and used uniquely designed serial BiFC assays to verify the essential role of Neck-domain for the ACE2/S protein PPI. We found that only the ACE2 fragment capable of self-dimerization can effectively bind to the S protein of SARS-CoV-2. This is consistent with the previous observation from a cryl-EM structural study (8). These data indicate that conformational changes caused by dimerization could significantly enhance the binding affinity of ACE2 to S protein. We further identified that the presence of Neck-domain is critical for successful ACE2/S protein recognition, which indicates that the disruption of the Neck-domain-mediated ACE2 dimerization in host cells might have a significant impact on blocking S protein recognition.
We further identified that the minimal peptide sequence from residues 616–658 (NKN1) in Neck-domain is necessary for blocking ACE2 dimerization and the PPI between ACE2 and S protein. It is interesting that the NKA-1 fragment (Figure 5A) containing both the second and forth helices could not effectively inhibit the PPI between ACE2 or ACE2/S protein. One reason could be the potential self-association of NKA-1 fragments caused by an extensive hydrogen bond and cation-π interaction among residues within these two helices (Figure 7A). By expressing NKN1 or NK-NT peptides in ACE2-expressing cells, we confirmed that these peptides could inhibit the S protein pseudotyped lentivirus infection. These results correlate well with a previous study showing that artificial dimerization of ACE2 PD by fusion with the IgG Fc has a much lower efficiency than that of ACE2-ECT in binding to S protein and blocking SARS-CoV-2 pseudovirus infection (17, 18). The data here allowed us to predict that, on the host cell surface, ACE2 receptors form a homodimer to enable S protein recognition, and the dimerization mediated by Neck-domain is necessary for ACE2/S protein interaction and the SARS-CoV-2 viral infection. Thus, drugs interfering with Neck-domain to disrupt ACE2 homodimerization would have the potential to minimize the susceptibility of host cells to SARS-CoV-2 virus.
As a transmembrane zinc metallocarboxypeptidase, ACE2 is abundantly expressed in many organs/tissues and promotes cardioprotective actions of nitric oxide release, vasodilation, and the inhibition of inflammation by cleaving angiotensin II (Ang II) into Ang-(1-7) and Ang I into Ang-(1-9) (19, 20). While the elevated level of ACE2 in patients with comorbidities is associated with an increased risk of SARS-CoV-2 infection and severity, the enzyme activity of ACE2 may be beneficial for patients suffering a dysfunctional renin–angiotensin–aldosterone system, which poses danger to infected patients with various comorbidities (21). Although evidence showing that ACE2 lacking Neck-domain maintains its peptidase activity (18), further investigation would be necessary to evaluate the potential impact of disturbing ACE2 dimerization to human health and to establish targeting the ACE2 dimerization as a potential therapeutic tool against SARS-CoV-2 infection.
We therefore expect that future research would identify refined protein peptides based on Neck-domain and/or small molecules that can effectively block the ACE2 dimerization to enhance the host cell “immunity” to SARS-CoV-2 virus. These drugs could be applied either to protect the host from viral infection or to curb subsequent viral amplification and viremia in infected hosts. This could also have an extended effect on preventing the infection by other members of the coronavirus family that utilize ACE2 receptor to enter host cells.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
YT and JZ conceived and designed the study and wrote the manuscript. JZ and YS performed the experiments, conducted data analysis, and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the University of Connecticut OVPR COVID-19 Rapid Start Funding, and the USDA National Institute of Food and Agriculture AFRI Project 2022-67016-37126.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181271-280:e8. doi: 10.1016/j.cell.2020.02.052
2. Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature (2020) 581:221–4. doi: 10.1038/s41586-020-2179-y
3. Datta PK, Liu F, Fischer T, Rappaport J, Qin X. SARS-CoV-2 Pandemic and Research Gaps: Understanding SARS-CoV-2 Interaction With the ACE2 Receptor and Implications for Therapy. Theranostics (2020) 10:7448–64. doi: 10.7150/thno.48076
4. Seyedpour S, Khodaei B, Loghman AH, Seyedpour N, Kisomi MF, Balibegloo M, et al. Targeted Therapy Strategies Against SARS-CoV-2 Cell Entry Mechanisms: A Systematic Review of In Vitro and In Vivo Studies. J Cell Physiol (2020) 236:2364–92. doi: 10.1002/jcp.30032
5. Tay MZ, Poh CM, Renia L, Macary PA, Ng LFP. The Trinity of COVID-19: Immunity, Inflammation and Intervention. Nat Rev Immunol (2020) 20:363–74. doi: 10.1038/s41577-020-0311-8
6. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM Structure of the 2019-Ncov Spike in the Prefusion Conformation. Science (2020) 367:1260–3. doi: 10.1126/science.abb2507
7. Walls AC, Park YJ, Tortorici MA, Wall A, Mcguire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell (2020) 181281-292:e6. doi: 10.1101/2020.02.19.956581
8. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science (2020) 367:1444–8. doi: 10.1126/science.abb2762
9. Kodama Y, Hu CD. An Improved Bimolecular Fluorescence Complementation Assay With a High Signal-to-Noise Ratio. Biotechniques (2010) 49:793–805. doi: 10.2144/000113519
10. Kanarek N, Keys HR, Cantor JR, LEWIS CA, CHAN SH, Kunchok T, et al. Histidine Catabolism Is a Major Determinant of Methotrexate Sensitivity. Nature (2018) 559:632–6. doi: 10.1038/s41586-018-0316-7
11. Hu CD, Chinenov Y, Kerppola TK. Visualization of Interactions Among bZIP and Rel Family Proteins in Living Cells Using Bimolecular Fluorescence Complementation. Mol Cell (2002) 9:789–98. doi: 10.1016/S1097-2765(02)00496-3
12. Bhat RA, Lahaye T, Panstruga R. The Visible Touch: In Planta Visualization of Protein-Protein Interactions by Fluorophore-Based Methods. Plant Methods (2006) 2:12. doi: 10.1186/1746-4811-2-12
13. Miller KE, Kim Y, Huh WK, Park HO. Bimolecular Fluorescence Complementation (BiFC) Analysis: Advances and Recent Applications for Genome-Wide Interaction Studies. J Mol Biol (2015) 427:2039–55. doi: 10.1016/j.jmb.2015.03.005
14. Huang C, Bernard D, Zhu J, Dash RC, Chu A, Knupp A, et al. Small Molecules Block the Interaction Between Porcine Reproductive and Respiratory Syndrome Virus and CD163 Receptor and the Infection of Pig Cells. Virol J (2020) 17:116. doi: 10.1186/s12985-020-01361-7
15. Frankel AD, Pabo CO. Cellular Uptake of the Tat Protein From Human Immunodeficiency Virus. Cell (1988) 55:1189–93. doi: 10.1016/0092-8674(88)90263-2
16. Tang Y, Lin CJ, Tian XC. Functionality and Transduction Condition Evaluation of Recombinant Klf4 for Improved Reprogramming of iPS Cells. Cell Reprogram (2011) 13:99–112. doi: 10.1089/cell.2010.0072
17. Li Y, Wang H, Tang X, Fang S, Ma D, Du C, et al. SARS-CoV-2 and Three Related Coronaviruses Utilize Multiple ACE2 Orthologs and Are Potently Blocked by an Improved ACE2-Ig. J Virol (2020) 94(22):e01283–20. doi: 10.1128/JVI.01283-20
18. Xiao T, Lu J, Zhang J, Johnson RI, Mckay LGA, Storm N, et al. A Trimeric Human Angiotensin-Converting Enzyme 2 as an Anti-SARS-CoV-2 Agent. Nat Struct Mol Biol (2021) 28:202–9. doi: 10.1038/s41594-020-00549-3
19. De bruin AM, Libregts SF, Valkhof M, Boon L, Touw ,IP, Nolte MA. IFNgamma Induces Monopoiesis and Inhibits Neutrophil Development During Inflammation. Blood (2012) 119:1543–54. doi: 10.1161/ATVBAHA.120.314558
20. Gustafson D, Raju S, Wu R, Ching C, Veitch S, Rathnakumar K, et al. Overcoming Barriers: The Endothelium As a Linchpin of Coronavirus Disease 2019 Pathogenesis? Arterioscler Thromb Vasc Biol (2020) 40:1818–29. doi: 10.3390/microorganisms9081692
Keywords: ACE2, S protein, dimerization, protein–protein interaction, peptides, BiFC, SARS-CoV-2
Citation: Zhu J, Su Y and Tang Y (2022) Disrupting ACE2 Dimerization Mitigates the Infection by SARS-CoV-2 Pseudovirus. Front. Virol. 2:916700. doi: 10.3389/fviro.2022.916700
Received: 09 April 2022; Accepted: 03 June 2022;
Published: 04 July 2022.
Edited by:
Jun Wang, The State University of New Jersey, United StatesReviewed by:
Krishna M. Padmanabha Das, Harvard Medical School, United StatesKrishnan Harshan, Centre for Cellular & Molecular Biology (CCMB), India
Copyright © 2022 Zhu, Su and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Young Tang, yong.tang@uconn.edu