
Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4


Department of Pharmaceutical Sciences, University of Perugia, via Fabretti, 06123 Perugia, Italy
Int. J. Mol. Sci. 2021, 22(13), 7135; https://doi.org/10.3390/ijms22137135
Submission received: 20 May 2021
/
Revised: 19 June 2021
/
Accepted: 29 June 2021
/
Published: 1 July 2021
(This article belongs to the Special Issue Neuroinflammation and Cell Death: What Is New?)
Abstract
:Parkinson’s disease (PD) is the most common neurodegenerative motor disorder characterized by selective degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) of the midbrain, depletion of dopamine (DA), and impaired nigrostriatal pathway. The pathological hallmark of PD includes the aggregation and accumulation α-synuclein (α-SYN). Although the precise mechanisms underlying the pathogenesis of PD are still unknown, the activation of toll-like receptors (TLRs), mainly TLR4 and subsequent neuroinflammatory immune response, seem to play a significant role. Mounting evidence suggests that viral infection can concur with the precipitation of PD or parkinsonism. The recently identified coronavirus named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of ongoing pandemic coronavirus disease 2019 (COVID-19), responsible for 160 million cases that led to the death of more than three million individuals worldwide. Studies have reported that many patients with COVID-19 display several neurological manifestations, including acute cerebrovascular diseases, conscious disturbance, and typical motor and non-motor symptoms accompanying PD. In this review, the neurotropic potential of SARS-CoV-2 and its possible involvement in the pathogenesis of PD are discussed. Specifically, the involvement of the TLR4 signaling pathway in mediating the virus entry, as well as the massive immune and inflammatory response in COVID-19 patients is explored. The binding of SARS-CoV-2 spike (S) protein to TLR4 and the possible interaction between SARS-CoV-2 and α-SYN as contributing factors to neuronal death are also considered.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
According to the World Health Organization (WHO), Parkinson’s disease (PD) is the second most common neurodegenerative disease in the world, after Alzheimer’s disease (AD), and one of the most common causes of neurological disability with a high social impact [1]. The prevalence of PD is about 10 million people worldwide, and it is estimated that there will be about 13 million people with PD by 2040 [2]. PD is a chronic, progressive, age-related neurodegenerative disease clinically characterized by motor symptoms, such as bradykinesia, rigidity, tremor at rest, slow movements, and postural instability [3]. Motor symptoms are the consequence of the progressive loss of neuromelanin-containing neurons, especially dopaminergic neurons (DA) in the substantia nigra pars compacta (SNpc) and dopamine depletion in the striatum, as confirmed by post-mortem study of clinically diagnosed PD patients [4,5]. The pathological hallmark of PD is represented by the accumulation of cytoplasmic inclusions such as Lewy bodies (LB) and Lewy neurites rich in α-synuclein (α-SYN) [6,7,8].
Albeit a number of genetic and environmental risk factors have been characterized, the cause(s) of PD are still unknown. In the last decades, studies have suggested the association between certain viral infections and acute and chronic parkinsonism. Most virus species include herpesvirus, Coxsackie, Japanese encephalitis B, Epstein Barr, Human Immunodeficiency Virus (HIV), and western equine encephalitis [9,10,11,12,13,14,15,16,17].
In the CNS, specialized innate immune sentinels, such as microglia, macrophages, dendritic cells, and astrocytes detect and clear viral agents by activating a robust immune response. However, many viruses use different strategies to elude immune system, cross the blood–brain barrier (BBB), and directly enter the nervous system. For example, viruses can enter the CNS via a “Trojan horse” mechanism using infected leucocytes as a vehicle for the passage from the blood through the BBB. [18]. Viruses may cause the disruption of the BBB integrity to gain access to the CNS [19]. Viral invasion of neural tissues can initiate inflammatory signaling by local primed immune cell recruitment, resulting in the release of abundant levels of an array of proinflammatory cytokines/chemokines that in turn can disrupt the BBB and increase its permeability [20].
The connection between systemic inflammation, PD, and neuroinflammation has been largely elucidated and a number of studies suggest that Toll-like receptors (TLRs), mainly TLR2 and 4, participate in the pathogenesis of PD as promoters of immune/neuroinflammatory responses that precede both motor and non-motor symptoms. The overexpression of TLR4 has been found in circulating monocytes of PD patients, in B cells, and in the caudate/putamen [21,22,23,24]. Studies in animal models of PD reported the potential role of TLR4 in mediating biochemical changes as well as dopaminergic cell death and α-synuclein accumulation in the midbrain [25,26]. Moreover, TLR4 has been found to play a critical role as a mediator of the neurotoxicity induced by α-synuclein oligomers [27].
Coronavirus disease (COVID-19) is an ongoing pandemic caused by a novel RNA (32 Kb genome) virus, namely, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), whose worldwide cases passed 160 million. COVID-19 deaths passed three million worldwide, and unfortunately these numbers are destined to further increase [28]. It is a highly transmissible virus strain of the recently discovered coronavirus in China and is transmitted primarily via respiratory droplets. It shares genetic identity with SARS-CoV and MERS-CoV [29,30]. It is composed of four major proteins named the spike (S), envelope (E), membrane (M), and nucleocapsid (N) [31]. All these proteins are notably implicated in viral infection, proliferation or host cell pathogenesis and are therefore predictable as the potential targets for vaccine or drug development.
SARS-CoV-2 acts through the binding between the viral receptor S protein and different glycoprotein receptors on the cell surface. The virus S protein is a flexible and instable glycosylated complex. Recently, high-quality and stabilized forms of the trimeric S protein have been produced for use in vaccines and diagnostic tests [32].
A priming step of virus infection is represented by the cleavage of S protein by the transmembrane serine protease 2 (TMPRSS2) [33]. Then, cellular endocytosis occurs and RNA replication stages can take place. The main protein target on the cell surface is the angiotensin converting enzyme 2 (ACE2), an enzyme involved in converting angiotensin II to angiotensin. ACE2 is highly expressed in a wide variety of human tissues, including the brain [34,35,36,37,38].
SARS-CoV-2 has potential affinity to other candidate functional receptors such as dipeptidyl peptidase 4 [39], sialic acid residues on surface of airway cells and neurons [40], and lectin CD209L [41].
The respiratory tract is the first site that can be infected by SARS-CoV-2. If the virus is not cleared by the immune response, it reaches the lower respiratory tract and rapidly can be transmitted through droplets in a human-to-human manner [42]. COVID-19 includes a wide spectrum of clinical manifestations ranging from respiratory illness, with symptoms such as bronchitis, pneumonia, asthma, chronic obstructive pulmonary disease, and severe respiratory distress syndromes to multiorgan severe inflammation [43,44]. The high levels of cytokines secreted during the multisystem inflammatory syndrome can cause septic shock, sometimes fatal for the patients. Importantly, host response is fundamental to challenge the uncontrolled inflammatory response and plays a crucial role in susceptibility to SARS-CoV-2-mediated diseases [45].
Epidemiologic studies demonstrate that, unlike polio and others virus that evocate more severe complications for the young, SARS-CoV-2 infection is a zoonosis that affects older people, suggesting that age-associated chronic inflammation may occur with the development of disease [46].
A higher pathogenicity and death rate have been observed in particularly vulnerable and frail subjects and/or those affected by chronic inflammation or multiple comorbidities including diabetes, obesity, cardiovascular diseases, and immunosuppression [47,48,49]. Recent genome-wide association studies identified potential genetic factors involved in the development of COVID-19 [50]. Although the precise factors determining SARS-CoV-2 infection are poorly understood, systemic immune and hyperinflammatory responses play a major role. For example, it has demonstrated that the interaction between ACE2 and the S protein stimulates NF-kB activation, which results in the release of proinflammatory cytokines in infected cells [51]. The cytokine storm affecting hospitalized patients with severe COVID-19 is responsible for multiple organ failure and includes the nervous system.
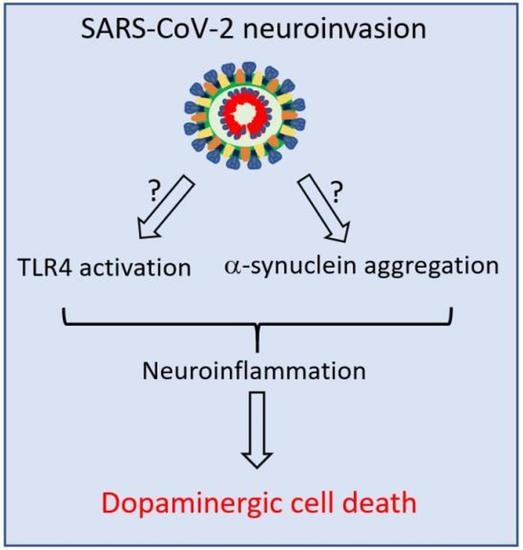
This review focuses on the suspected intriguing link between SARS-CoV-2 infection and the pathogenesis of PD with particular attention on the potential involvement of TLR4 as a common pathway during harmful inflammatory responses in both diseases.
Although TLR4 signaling represents one of a series of immune and inflammatory factors found activated during both PD and COVID 19, the strong interaction of TLR4 with the SARS-CoV-2 S protein as well as the possible binding between SARS-CoV-2 and α-SYN suggest triggering mechanisms of neurodegenerative processes underlying PD, which deserve to be elucidated.
2. Neuroinflammation
Neuroinflammation is a complex process able to activate the innate immune response in the central nervous system (CNS), associated not only with neurodegenerative diseases but also with infections and trauma. For a long time, the CNS has been considered an immunologically privileged site because the lack of cells capable of presenting the antigen in the brain parenchyma, the absence of lymphatic drainage, and the presence of BBB. Over the last 30 years, this concept has been challenged, and many reports show active cooperation between the CNS and immune system despite the physical barrier [52,53].
A growing body of evidence indicates that neuroinflammation is involved in the pathogenesis of several neurodegenerative diseases including PD [54,55,56,57], Alzheimer’s disease (AD) [58], amyotrophic lateral sclerosis [59], Huntington’s disease, and virus-associated dementia [60,61]. Neuroinflammation implicates the recruitment of immune and non-immune cells at the site of injury or infection, such as resident macrophages, i.e., microglia, aside from astrocytes, endothelial cells, infiltrating T-lymphocytes, α-SYN reactive T-lymphocytes, and major histocompatibility complex (MHC) class II-positive microglia [62,63,64,65]. Microglia are the most abundant populations in the brain parenchyma and represent the major cell type involved in neuroinflammation However, sophisticated neuron–microglia–astrocyte crosstalk also plays a critical role in the immune response [66,67,68].
Although no standardized morphological classification exists, the mature brain microglial cells are generally quiescent or “resting” and exhibit a ramified shape with fine extending protrusions. They also are equipped with a wide range of receptors, comprising colony-stimulating factor 1 receptors that continuously scan the surrounding environment and control important functions such as synaptic activity, clearance of cell debris, and cerebral homeostasis [69,70,71,72].
Immunological and inflammatory stimuli or invading agents, including viruses, can promote phenotype switching between a highly ramified morphology and a large ameboid-like shape with short branches typical of activated microglia [73,74,75]. This switching triggers a complex cascade of events whose magnitude depends on the intensity and duration of the stimulation [76].
While acute insults evoke protection and healing of the brain tissue, chronic stimulation causes a sustained inflammatory response resulting from the acquisition of an array of functions by microglia associated with upregulation of a large number of receptor types, such as cytokine receptors, TLRs or cell adhesion molecules.
The activation of TLRs initiates intracellular signaling pathways that culminate with the release of interleukins, interferons, chemokines, and other toxic compounds. Chemokines attract more microglia that further contribute to propagate the neuroinflammatory process and activate the apoptotic machinery [77].
Whether neuroinflammation is a consequence or a cause of nigral cell loss in PD is still unclear. Certainly, several neurodegenerative diseases, including PD, are known to impact the switch of microglia from the neuroprotective to the neurotoxic phenotype [78,79,80,81]. Sustained gliosis seems to be the prominent pathological feature of PD that is associated with loss of dopaminergic neurons in the SNpc [82,83,84]. Importantly, the degeneration of the dopaminergic neurons could be caused not only by glial response but also by the loss of a supporting role by resting microglia following switching to an activated status. Neuroinflammation in PD is also accompanied by microglia uptake and phagocytosis of neuromelanin released by degenerated DA neurons into the extracellular compartment that contribute to the acceleration of the neurodegenerative process.
In 1988, McGeer et al. [85] showed the presence of reactive microglia in the SNpc of human post-mortem brain tissues. High levels of interleukin-1β (IL1-β), IL-6, TNFα, interferon γ (IFNγ), cyclooxygenase type 2 (Cox-2), nitric oxide synthase (NOS), infiltrating peripheral immune cells, and ROS were found in post-mortem PD brains as well as in cerebrospinal fluids of patients with PD. A recent postmortem study on demented PD cases revealed the upregulation of neuroinflammatory markers and stated that α-SYN pathology and microglia activation play a critical role [86,87].
3. Toll-Like Receptors
TLRs are type I transmembrane glycoproteins with an extracellular leucine-rich repeat motif and a cytoplasmic Toll/IL-1 receptor (TIR) signaling domain, similar to the interleukin-1 receptor domain (IL-1R) [88]. Both IL-1R and TLRs initiate downstream signaling, leading primarily to the activation of the transcription factor NF-kB, a key regulator of inflammatory response [89]. TLRs belong to the complex pattern recognition receptors (PRRs) expressed in immune and non-immune cells, including neurons and glia, which are involved in regulating the innate immune system and inflammatory response by producing inflammatory cytokines and other mediators [90]. These processes prime immediate host-defense responses crucial for the clearance of infecting agents and for the following adaptative immune responses [91].
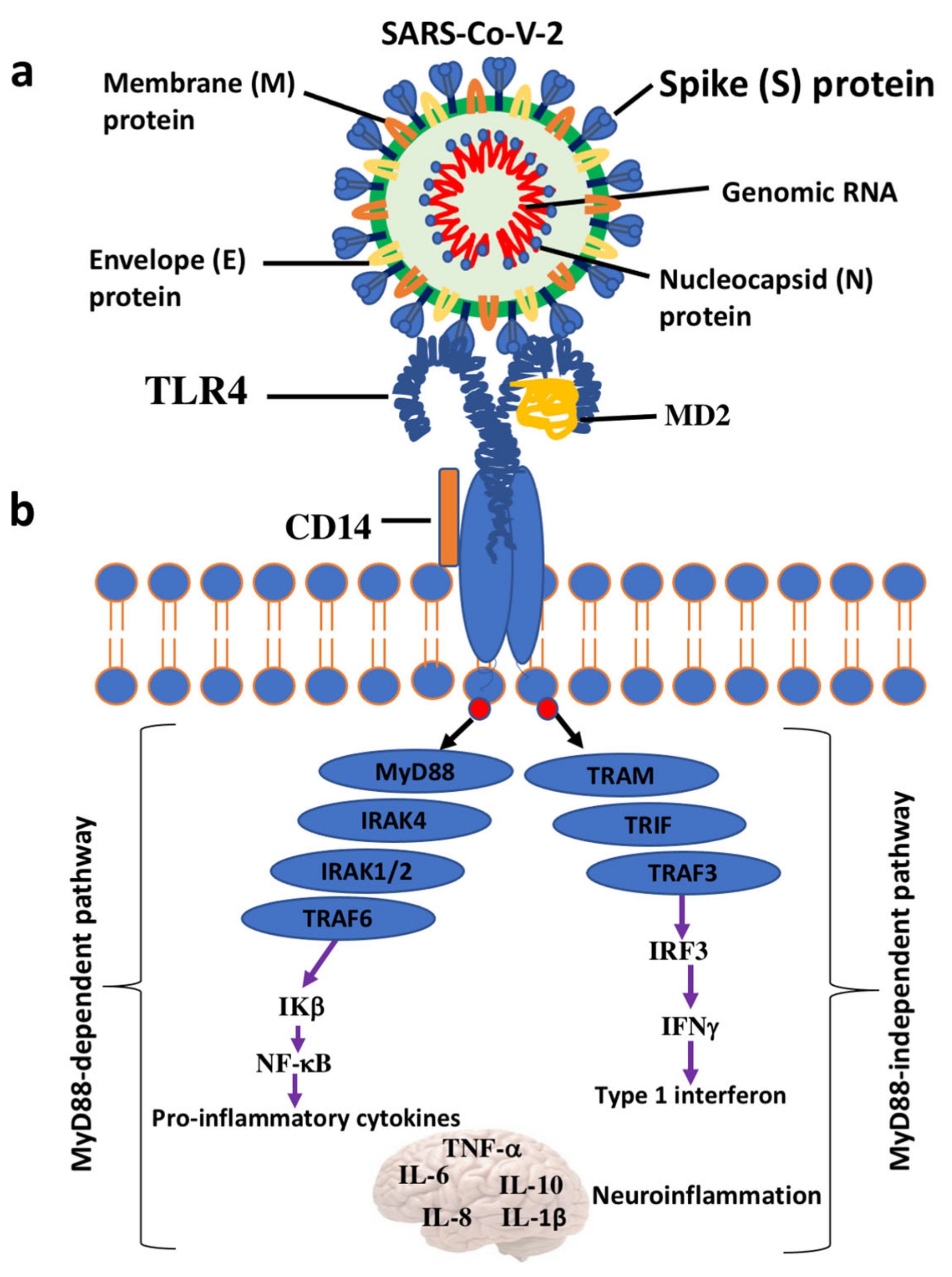
TLRs are highly specialized in sensing invading bacteria, viruses, parasites, and cell debris [92,93]. The TLR family comprises 11 members (TLR1–TLR11) in human and 12 (TLR1–TLR9, TLR11–TLR13) in mouse. They are localized on the cell surface as well as in intracellular compartments such as the endoplasmic reticulum, endosome, lysosome, or endolysosome, which normally respond to viral nucleic acids. However, they survey for the presence of structural motifs in a wide array of invading microorganisms, named pathogen-associated molecular patterns (PAMPs), in the extracellular space and within endocytic compartments. TLRs also sense endogenous damage or danger molecular patterns (DAMPs), also known as alarmins, released by damaged cells and injured tissues or derived from apoptotic and necrotic cells [93]. The activation of neuro-inflammatory machinery starts with the binding between DAMPs/PAMPs and TLRs and advances with the dimerization of TLRs and the subsequent interaction with adaptor proteins such as Myeloid Differentiation Primary Response Gene 88 (MyD88) and Toll/interleukin-1 receptor-like (TIR)-domain containing adapter-inducing interferon-β (TRIF) domain. This cascade of events leads to the recruitment of others complexes, such as IL-1R-associated kinase (IRAK) and MAP kinases, and the nuclear translocation/activation of transcription factors, including NF-kB, that ultimately trigger the downstream overexpression of pro-inflammatory genes and cell degeneration [94] (Figure 1). Among the 11 human TLRs members, TLR4 seems to play a critical role in the development and progression of neurodegenerative diseases, including PD [95]. A broad variety of molecules are recognized by TLR4. They include (1) exogenous natural ligands such as canonical lipopolysaccharide (LPS) from gram-negative bacteria, viruses, fungi, and mycoplasmas); (2) extracellular matrix ligands (hyaluronan, biglycan, fibronectin, and heparan sulphate); (3) intracellular and secreted endogenous ligands (heat shock proteins, defensins, S100 proteins, and amyloid β) [96].
4. TLR4
Analysis of transcriptomic data from human postmortem control brains reveals ubiquitous expression of TLR4 throughout the brain, with their expression being higher in the substantia nigra and the putamen. Increased TLR4 protein levels were found in peripheral immune cells as well as in the substantia nigra and caudate/putamen of PD cases [24,97].
Additional studies in experimental models of PD and α-synucleinopathies demonstrated the important role of TLR4 in α-synucleinopathies and the constitutive expression in microglia and the up-regulation in the substantia nigra [68,69,97,98,99,100].
TLR4 is expressed by two types of non-neuronal supportive cells: the CNS residential macrophages or microglia and the macroglial cells such as astrocytes.
In the brain, TLR4-mediated signaling pathways have been implicated in the pathogenesis of PD [103]. Its stimulation causes the production of elevated levels of neurotoxic inflammatory cytokines that damage nigral dopaminergic neuron [104]. α-SYN as well as neuromelanin and other molecules released by damaged neurons can act as ligands for TLRs, including TLR4, and generate a sustained immune response that initiates inflammatory processes implicated in PD [105,106].
Drouin-Ouellet and co-workers [23] showed that the overexpression of α-SYN induced a critical modulation of TLR4 signaling in the blood and brain of both experimental models and PD patients. This triggers an immune/inflammatory response that culminates with neuronal death and negatively impacts motor symptoms. However, a study supported a role for TLR4 in mediating the clearance of α-SYN, suggesting an innate neuroprotective mechanism [107]. Therefore, both neuroprotective and detrimental roles of TLR4 in PD have been suggested: acute stimuli such as posttranslational modification of α-SYN can be a trigger for TLR4 microglial activation and protein clearance. Conversely, chronic inflammation, from inside or outside the nervous system, may promote the imbalanced activation of TLR4 signaling and escalation of inflammatory response, which may contribute to the pathogenesis of PD [108,109]. A large number of molecules, including several forms of α-SYN (monomer, oligomers, and fibrils, truncated and phosphorylated), released from neighbouring neurons in the extracellular milieu, act as ligands for TLR4 and undergo phagocytosis, degradation, and clearance. If comprised, this mechanism can lead to further inflammatory signals that occur with the formation of aggregated forms of α-SYN in the Lewy bodies of substantia nigra.
The formation of the complex TLR4-myelod differentiation 2 (MD2) on the cell surface, as well as the presence of LPS-binding proteins and cluster of differentiation 14 (CD14), is required for the recognition of ligands and the activation of intracellular pathways. The dimerization of TLR4-MD2 induces conformational changes that endorse the recruitment of adaptor proteins containing Toll/interleukin-1 receptor-like (TIR) domains. This is essential to start two possible intracellular signal pathways: the MyD88-dependent pathway culminating with the release of proinflammatory cytokines and the MyD88-independent pathway leading to the production of the type I interferons [110].
The engagement of MyD88 and MyD88-adaptor-like (MAL) protein induces a cascade of downstream events including the phosphorylation of IL-1R-associated kinases (IRAKs), the association of TNF-receptor-associated factor 6 (TRAF6), and the activation of transforming growth factor β-activated kinase 1 (TAK1). Many other adaptor proteins are involved and include TAK1-binding protein 2 (TAB2) and TAK1-binding protein 3 (TAB3). TAK1, in turn, activates two different kinase types: the mitogen-activated protein kinases (MAPKs), which comprise JUN N-terminal kinase (JNK), p38, extracellular signal-regulated kinases (ERK1/2), and the IkB kinase complex (IKK). The final response is the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and activator protein-1 and the abundant secretion of proinflammatory cytokines [111]. Instead, the activation of the MyD88-independent pathway is started by internalization of the TLR4-MD2 complex by CD14 and the recruitment/activation of adaptor proteins TRIF, TRIF-related adaptor molecule (TRAM), and TRAF3 followed by IFN regulatory factor 3 (IRF3) nuclear translocations. These events culminate with the production of type I IFNs [112]. Sustained activation of these signals attracts macrophages, natural killer, and other cells and may result in the toxic accumulation of ROS and RNS in different cell types, including neuronal cells [113,114] (Figure 1).
5. SARS-CoV-2 Inflammatory Response: Role of TLRs
Currently, the identification of the precise mechanism underlying the cytokine storm in hospitalized patients with severe COVID-19 is essential. The severity of COVID-19 is associated with the excessive inflammatory innate response and dysregulated adaptive host immune defense. In this context, TLRs can have a dual role. In fact, the activation of the innate immune system through TLRs can be the first line of defense against invading viruses and can support the elimination of viruses. However, prolonged and dysregulated activation may contribute to the onset of the hyperinflammation and poor outcome of clinical manifestations of COVID-19 [115,116].
Research on influenza viruses suggests that the expression of cytokines by lung epithelial, macrophages, and dendritic cells is the result of the activation, by viral PAMPs, of intracellular or extracellular TLRs. The main viral PAMPs are proteins, double strand RNA (dsRNA), single strand RNA (ssRNA), and CpG DNA, which are recognized by distinct TLRs, including TLR2, TLR3, TLR7, TLR8, and TLR9 [117]. Upon ligand recognition, all TLRs except TLR3 recruit MyD88. TLR3 and TLR4 also recruit the adapter protein TRIF. The MyD88-dependent and TRIF cascades initiate the nuclear translocation of NF-κB, IRF3, and IRF7 transcription factors that culminate with the upregulation of inflammatory cytokines/chemokines (through NF-kB) and IFN genes (through both IRF3 and IRF7) [118].
Systemic inflammation in patients with COVID-19 has been associated with elevated serum levels of pro-inflammatory cytokines and chemokines such as IL-2, IL-7, IL-8, IL-10, IP-10 (interferon-γ-inducible protein), MCP-1 (monocyte chemoattractant protein), MIP-1α (macrophage inflammatory protein 1 alpha), and TNFα. Additionally, high levels of IgG and a high neutrophil-to-lymphocyte ratio have been observed [116,119].
Recently, TLRs signaling pathways have been recognized as accessory factors that may occur with the COVID-19 pathogenesis. The main members include TLR2, TLR3, TLR4, TLR6, TLR7, TLR8, and TLR9, with beneficial and harmful effects towards SARS-CoV-2 infection. The activation of TLR7/8 has been found to evoke a strong pro-inflammatory response during acute lung injury [120]. Furthermore, the binding of SARS-CoV-2 PAMPs to the extracellular domain of human TLR1, TLR4, and TLR6 seems to be crucial for COVID-19 immunopathogenesis [121,122].
The interaction between the SARS-CoV-2 S protein and cell surface TLRs has been investigated by molecular docking studies. The results have demonstrated a significant binding of the S protein to TLR1, TLR4, and TLR6, especially TLR-4, suggesting a potential mechanism of the cytokine cascade [121].
6. Interaction between the SARS-CoV-2 S protein and TLR4
Although the host genetic background powerfully influences the requirement for TLR4-mediated signals during SARS-CoV-2 infection, TLR4 activation seems to represent one of the critical steps for host–virus general interaction [122]. It has been demonstrated that TLR4 deficiency can induce resistance to acute manifestations that accompany the viral attack, suggesting an important role of TLR4 signaling in the profuse pro-inflammatory cytokine storm [123].
Structural analysis by molecular docking revealed that the strongest binding was observed between the native spike glycoprotein and human TLR4 compared with other TLRs present in the human cells [121]. The spike protein is a surface-exposed homo-trimeric transmembrane, heavily glycosylated complex with high binding affinity for the ACE2 receptor of human host cells 32. Another recent study found an interaction between the S protein and Escherichia coli LPS, the well-known activator of TLR4 [124].
A robust binding was also observed with the TLR4 accessory proteins CD14 and MD-2 that are overexpressed in inflammatory circumstances [125]. Interestingly, multi-epitopic regions of the SARS-CoV-2 S protein have been found to interact with the TLR4/MD2 complex. Therefore, it could act as a potent peptide vaccine candidate against COVID-19 [125]. Studies using surface plasmon resonance confirm the direct binding between the SARS-CoV-2 trimer and TLR4 in THP-1 cell lines, neutrophils, primary bone marrow-derived macrophages, and peritoneal macrophages [126]. Ziegler et al. [127] showed that the interaction between the S protein and TLR4 induced the overexpression of interferon-stimulated genes, which in turn provoked the upregulation of ACE2. By this ingenious strategy, the viral entry is facilitated.
Although requiring confirmation, hydrogen bonds and hydrophobic interactions seem to assist the formation of the TLR4-spike complex. Together with ACE2, this event may be crucial for the activation of TLR4 signaling, NF-kB translocation, and hyperinflammation accompanied by the release of the TNF-α, IL-6 cytokines and more robustly IL1β [128]. Further studies are necessary to identify the specific TLR4 motifs involved. No interaction was observed by using the N-terminal or the receptor-binding domains of the spike protein [129] (Figure 2).
7. TLR4 and PD Pathogenesis
Both COVID-19 and PD are associated with dysfunction of the immune system and neuroinflammation, although the precise mechanisms remain unknown. The overexpression of TLR4 in PD and in COVID-19 patients together with high levels of many other inflammatory mediators could explain the link between these diseases. TLR4 downstream signaling molecules and inflammasome-related proteins were found to be upregulated in the blood cells from COVID-19 and PD patients [130,131]. The binding of the SARS-CoV-2 S protein to TLR4 on the neuron surface could represent a trigger for neurodegeneration, especially for the dopaminergic neurons in the nigrostriatal system, which express TLR4. As result, the recruitment of intracellular signaling pathways, such as MYD-88 together with upregulation of inflammatory signaling molecules could lead to the NF-kB-induced release of cytokines, reactive oxygen species (ROS), and reactive nitrogen species (RNS), and finally culminate with neurodegeneration [132].
Dysregulation of TLR4 signaling has been shown to play a role in the initiation and/or progression of PD. Notably, a number of DAMPs are released as a result of neuronal injury in PD, such as misfolded and aggregated α-SYN. The latter alters TLR4 expression and can act as a TLR4 agonist, further exacerbating the inflammatory status through activation of surrounding neurons and microglia cells [133].
The increase in systemic TLR4 agonists such as oxidized lipoproteins and phospholipids, heat shock proteins, extracellular matrix protein, and many other molecules secreted during COVID-19 can act as PAMPs for TLR4 and contribute to further damage the host brain parenchyma. Overall, the permanence of the virus for a long time in the CNS could be a factor predisposing patients to neurodegenerative processes as in PD.
8. Neuroinvasive Potential of SARS-CoV-2 and PD Pathogenesis
The concept of “Neuro-COVID-19” is being mentioned increasingly, but whether neurological manifestations in patients with COVID-19 are due to direct invasion of the virus or result from SARS-CoV-2-dependent neuroinflammatory response remains speculative.
Historically, viral infections were associated with parkinsonism. Viral agents can act as an initiator of parkinsonism as well as of other neurological complications [134]. For example, viruses such as influenza, Coxsackie, Japanese encephalitis B, western equine encephalitis, herpes, and viruses related to acquired immunodeficiency disorder, including HIV, are associated with both acute and chronic parkinsonism [10,135]
The trans-synaptic transfer of CoV and avian bronchitis viruses, as well as of other respiratory viruses, has been widely documented [136,137]. Evidence showed the ability of the SARS virus to penetrate into the CNS and cause neuropathy and gliopathy [138]. SARS genome sequences were detected in the hypothalamus and cortex of SARS autopsies with clear signs of edema and neurodegeneration [139].
The respiratory distress caused by SARS-CoV-2 infection that emerged in December 2019 occurs in about 73% of COVID-19 hospitalized patients by a number of neurological signs associated with dysfunction of the central nervous system [140], peripheral, and enteric [141] nervous systems. The symptoms include olfactory, gustatory, dizziness, headache, dizziness, confusion, encephalitis, stroke, anorexia, myalgias, and gastrointestinal disorders such as nausea and vomiting. Severe COVID 19 patients under intensive treatment exhibit cerebrovascular disease, consciousness state, and skeletal muscle injury [142,143].
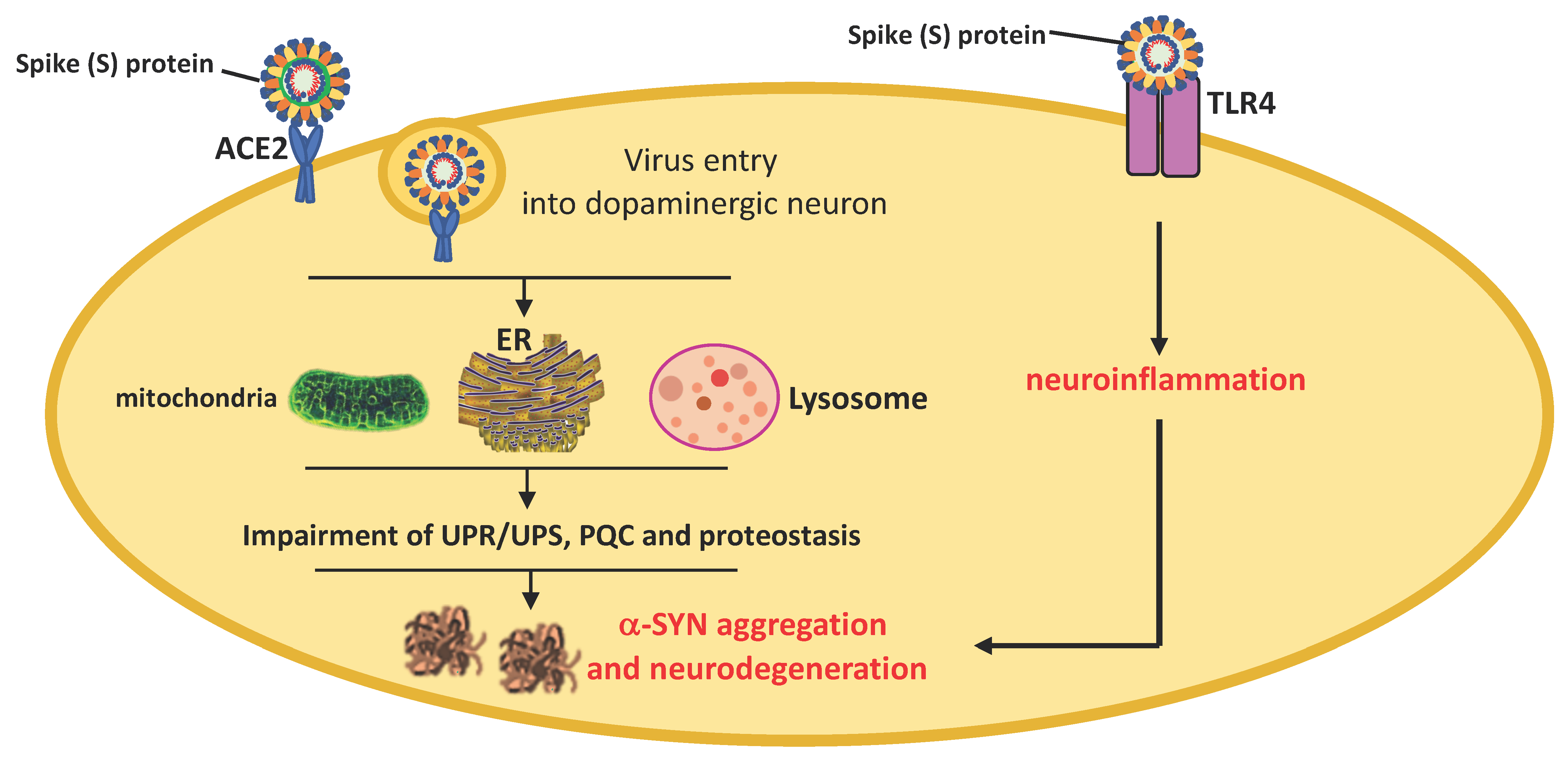
SARS-CoV-2 infects cells through the interaction between its S protein previously cleaved by TMPRSS2 and ACE2. The neuroinvasive propensity of SARS-CoV-2 is probably facilitated by high expression levels of ACE2 and TMPRSS2 in neurons, astrocytes, and oligodendrocytes that, together with TLR4 activation, can predispose patients to α-SYN aggregation, neurodegeneration, and PD pathogenesis [144]. As shown in the Allen Human Brain Atlas, ACE2 mRNA is widely distributed throughout the brain with notable strong expression in areas such as the cortex, striatum, hypothalamus, medulla, and substantia nigra [145]. ACE2 receptors are expressed in neurons, astrocytes, and oligodendrocytes in the substantia nigra and olfactory bulb [146]. However, ACE2 may not be the only binding site involved in brain uptake [147].
SARS-CoV-2 can circumvent the host immune response and spread within the CNS directly through the olfactory nerves [148] or similar to other viruses, can reach the brainstem through the vagus nerve, infect neurons of different areas, and induce a marked systemic pro-inflammatory response associated with the overproduction of cytokines [149,150,151,152,153]. Using a human brain organoid model, Song et al. [148] showed that SARS-CoV-2 can infect cells of neural origin and cause death of nearby cells. SARS-CoV-2 particles were also detected in the cortical neurons from postmortem autopsies of patients who died from COVID-19 as well as in the cerebrospinal fluid of SAR-CoV-2-positive cases [154].
The impact that COVID-19 might have on PD pathogenesis is debated at present. PD is associated with changes in the CNS immune response, microglia and oligodendroglial activation, upregulation of histocompatibility class II, and pro-inflammatory cytokine overproduction. SARS-CoV-2 infection may be a predisposing factor for an abundant immune response. The cytokine storm during COVID-19 can cause the breakdown of the BBB and lead to virus entry and immune cell infiltration [155]. This directly can cause neuronal death and escalation of patient care towards severe neurological complications characterizing all synucleinopathies, including PD [156,157,158,159] (Figure 3).
Parkinsonism has been reported following COVID-19. Although Parkinson’s disease was not diagnosed, functional nigrostriatal neuroimaging was abnormal in some COVID-19 cases, therefore presuming dopaminergic nigrostriatal impairment [160,161].
Moreover, some of the most common non-motor symptoms of PD are evident in COVID-19 patients and comprise anosmia/iposmia, gastrointestinal symptoms, ageusia, fatigue, and painful limbs [162,163]. These symptoms antedate the pathological deposition of α-SYN and the appearance of motor symptoms.
The possible trans-synaptic spread via the olfactory, lingual, and glossopharyngeal nerves of SARS-CoV-2 could explain the featured symptoms such as hyposmia and ageusia in many COVID-19 patients [164,165].
The presumed degeneration of dopaminergic nigrostriatal nerves could derive from immune activation in the olfactory system (without direct virus entry) that, associated with other toxic stress and the inability to activate neuroprotective responses, might promote α-synuclein misfolding, aggregation, and neurodegeneration.
Similar to PD, increased levels of IL-6 have been also found in COVID-19 patients, indicating an impact of inflammation on the progression of non-motor impairment [166,167,168].
α-SYN is the most important protein implicated in PD. Similar to West Nile virus and SAR-CoV-1, SARS-CoV-2 infection could lead to α-SYN upregulation in attempts to prevent viral replication and neuroinvasion [169,170]. However, abundant and protracted systemic inflammation could disturb the host cell proteostasis and protein quality control system, contributing to the pathological modification of α-SYN that can evolve with the formation of fibrillar structures. Although confirmation is needed, recent findings suggest that the interaction between the SARS-CoV-2 N protein and α-SYN could accelerate the protein aggregation into amyloid fibrils, leading to propagation and widespread neurodegeneration [171] (Figure 4).
Furthermore, α-SYN aggregation could also arise from the interaction between SARS-CoV-2 and damaged proteins belonging to autophagy machinery or involved in the maintenance of proteostasis. SARS-CoV-2 was reported to directly bind the human ORF8 protein, causing dysregulation of endoplasmic reticulum trafficking and aberrant proteostasis. This hazardous interaction could be crucial for pathological α-SYN aggregation/accumulation and could be a hindrance for its clearance [172,173,174,175,176] (Figure 5).
Finally, it was recently demonstrated that the expression of ACE2 as well as of other molecules that act as receptors for SARS-CoV-2, including CD209L and sialic acid residues, may be enhanced by interferons and other proinflammatory mediators released following viral infection [34]. Moreover, the interaction between viral proteins and extracellular host proteins can induce conformational changes and accelerate protein aggregation.
9. Conclusions
In conclusion, in this review, the possible impact of SARS-CoV-2 infection on the pathogenesis of PD has been discussed. Specifically, the involvement of TLR4 in mediating innate neuroinflammation in the COVID-19 pandemic and subsequent neurodegeneration has been hypothesized. The neuroinvasive potential of SARS-CoV-2 together with the capability to strongly interact with TLR4 through the SARS-CoV-2 S protein suggests an intriguing role for this receptor. A prolonged and dysregulated activation of TLR4 signaling, aside from the possible direct interaction between the SARS-CoV-2 N protein and α-SYN, may impair the protein quality control machinery and host cell proteostasis. The acceleration of the α-SYN aggregation into pathological multimeric protein species such as oligomers/protofibrils increases the vulnerability of nigrostriatal dopaminergic neurons, especially in older people, and leads to widespread neurodegeneration.
Certainly, the long-term link between SARS-CoV-2 infection and PD will be demonstrated when a number of postmortem studies are performed. Moreover, future follow-up of large cohorts of patients with COVID-19 and COVID-19 survivors will be useful to clarify the impact on PD onset and other neurodegenerative disorders.
Although speculative, the role of TLR4 signaling in the COVID-19 pandemic as the fil rouge for the detrimental interconnection between neuroinflammation, α-synuclein accumulation, and dopaminergic cell death deserves to be investigated.
Funding
This research received no external funding.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s disease. Subcell Biochem. 2012, 65, 389–455. [Google Scholar] [CrossRef] [Green Version]
- Dorsey, E.R.; Bloem, B.R. The Parkinson pandemic—A call to action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Moustafa, A.A.; Chakravarthy, S.; Phillips, J.R.; Gupta, A.; Keri, S.; Polner, B. Motor symptoms in Parkinson’s disease: A unified framework. Neurosci. Biobehav. Rev. 2016, 68, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar] [CrossRef]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, W.L.; Stott, K.; He, X.; Wood, T.; Yang, S.; Kwok, J.; Hall, K.; Zhao, Y.; Tietz, O.; Aigbirhio, F.; et al. Systemic α-synuclein injection triggers selective neuronal pathology as seen in patients with Parkinson’s disease. Mol. Psychiatry 2021, 26, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Big, C.; Reineck, L.A.; Aronoff, D.M. Viral infections of the central nervous system: A case-based review. Clin. Med. Res. 2009, 7, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Boltz, D.A.; Webster, R.G.; Smeyne, R.J. Viral parkinsonism. Biochim. Biophys. Acta 2009, 1792, 714–721. [Google Scholar] [CrossRef] [Green Version]
- Swanson, P.A.; McGavern, D.B. Viral diseases of the central nervous system. Curr. Opin. Virol. 2015, 11, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Bookstaver, P.B.; Mohorn, P.L.; Shah, A.; Tesh, L.D.; Quidley, A.M.; Kothari, R.; Bland, C.M.; Weissman, S. Management of viral central nervous system infections: A primer for clinicians. J. Cent. Nerv. Syst. Dis. 2017, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Olsen, L.K.; Dowd, E.; McKernan, D.P. A role for viral infections in Parkinson’s etiology? Neuronal. Signal. 2018, 2, NS20170166. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Yamada, T.; Nakajima, S.; Nakajima, K.; Yamamoto, T.; Okada, H. The substantia nigra is a major target for neurovirulent influenza A virus. J. Exp. Med. 1995, 181, 2161–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Limphaibool, N.; Iwanowski, P.; Holstad, M.J.V.; Kobylarek, D.; Kozubski, W. Infectious etiologies of parkinsonism: Pathomechanisms and clinical implications. Front. Neurol. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory response in the CNS: Friend or foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, M.D.; Salimi, H.; Gong, Y.; Yang, L.; Hamilton, S.L.; Heffernan, J.R.; Hou, J.; Miller, M.J.; Klein, R.S. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 2017, 308, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Najjar, S.; Najjar, A.; Chong, D.J.; Pramanik, B.K.; Kirsch, C.; Kuzniecky, R.I.; Pacia, S.V.; Azhar, S. Central nervous system complications associated with SARS-CoV-2 infection: Integrative concepts of pathophysiology and case reports. J. Neuroinflamm. 2020, 17, 231. [Google Scholar] [CrossRef]
- Rocha, N.P.; de Miranda, A.S.; Teixeira, A.L. Insights into neuroinflammation in Parkinson’s disease: From biomarkers to anti-inflammatory based therapies. Biomed. Res. Int. 2015, 628192. [Google Scholar] [CrossRef]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018, 4784268. [Google Scholar] [CrossRef] [Green Version]
- Refolo, V.; Stefanova, N. Neuroinflammation and glial phenotypic changes in alpha-synucleinopathies. Front. Cell Neurosci. 2019, 13, 263. [Google Scholar] [CrossRef] [Green Version]
- Drouin-Ouellet, J.; St-Amour, I.; Saint-Pierre, M.; Lamontagne-Proulx, J.; Kriz, J.; Barker, R.A.; Cicchetti, F. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int. J. Neuropsychopharmacol. 2014, 18, pyu103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.D.J.; Lucassen, P.J.; van Dam, A.M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014, 2, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariucci, G.; Pagiotti, R.; Galli, F.; Romani, L.; Conte, C. The potential role of toll-like receptor 4 in mediating dopaminergic cell loss and alpha-synuclein expression in the acute MPTP mouse model of Parkinson’s disease. J. Mol. Neurosci. 2018, 64, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Conte, C.; Roscini, L.; Sardella, R.; Mariucci, G.; Scorzoni, S.; Beccari, T.; Corte, L. Toll like receptor 4 affects the cerebral biochemical changes induced by MPTP treatment. Neurochem. Res. 2017, 42, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Gubernatorova, E.O.; Gorshkova, E.A.; Polinova, A.I.; Drutskaya, M.S. IL-6: Relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev. 2020, 53, 13–24. [Google Scholar] [CrossRef]
- Alexaki, A.; Wigdahl, B. HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. PLoS Pathog. 2008, 4, e1000215. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Pino, P.; Kint, J.; Kiseljak, D.; Agnolon, V.; Corradin, G.; Kajava, A.V.; Rovero, P.; Dijkman, R.; den Hartog, G.; McLellan, J.S.; et al. Trimeric SARS-CoV-2 spike proteins produced from CHO cells in bioreactors are high-quality antigens. Processes 2020, 8, 1539. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lazartigues, E. Expression of ACE2 in human neurons supports the neuro-invasive potential of COVID-19 Virus. Cell. Mol. Neurobiol. 2020, 1–5. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 virus targeting the CNS: Tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 14, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; McCord, K.A.; Bui, D.T.; Bouwman, K.M.; Kitova, E.N.; Kumawat, D.; Daskhan, G.C.; Tomris, I.; Han, L.; Chopra, P.; et al. Sialic acid-dependent binding and viral entry of SARS-CoV-2. BioRxiv 2021. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Geng, X.; Tan, Y.; Li, Q.; Xu, C.; Xu, J.; Hao, L.; Zeng, Z.; Luo, X.; Liu, F.; et al. New understanding of the damage of SARS-CoV-2 infection outside the respiratory system. Biomed. Pharmacother. 2020, 127, 110195. [Google Scholar] [CrossRef]
- Kalil, A.C.; Thomas, P.G. Influenza virus-related critical illness: Pathophysiology and epidemiology. Crit. Care 2019, 23, 258–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; McAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Denis, A.H.; Ross, O.A. Age and age-related diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Costa, F.F.; Rosário, W.R.; Ribeiro Farias, A.C.; de Souza, R.G.; Duarte Gondim, R.S.; Barroso, W.A. Metabolic syndrome and COVID-19: An update on the associated comorbidities and proposed therapies. Diabetes Metab. Syndr. 2020, 14, 809–814. [Google Scholar] [CrossRef]
- Del Turco, S.; Vianello, A.; Ragusa, R.; Caselli, C.; Basta, G. COVID-19 and cardiovascular consequences: Is the endothelial dysfunction the hardest challenge? Thromb. Res. 2020, 196, 143–151. [Google Scholar] [CrossRef]
- Roberts, K.A.; Colley, L.; Agbaedeng, T.A.; Ellison-Hughes, G.M.; Ross, M.D. Vascular Manifestations of COVID-19-Thromboembolism and Microvascular Dysfunction. Front. Cardiovasc. Med. 2020, 7, 598400. [Google Scholar] [CrossRef]
- The Severe Covid-19 GWAS Group. Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.Y.; Plociennikowska, A.; Heigwer, F.; Joecks, S.; Burkart, S.S.; Zander, D.Y. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-kB. BioRxiv 2020. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Kivisäkk, P.; Kidd, G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003, 3, 569–581. [Google Scholar] [CrossRef]
- Chavan, S.S.; Pavlov, V.A.; Tracey, K.J. Mechanisms and therapeutic relevance of neuro-immune communication. Immunity 2017, 46, 927–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.C.; Standaert, D.G. Ten unsolved questions about neuroinflammation in Parkinson’s disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef]
- Riederer, P.; Berg, D.; Casadei, N.; Cheng, F.; Classen, J.; Dresel, C.; Jost, W.; Krüger, R.; Müller, T.; Reichmann, H.; et al. α-Synuclein in Parkinson’s disease: Causal or bystander? J. Neural Transm. 2019, 126, 815–840. [Google Scholar] [CrossRef]
- Jellinger, K.A. Is Braak staging valid for all types of Parkinson’s disease? J. Neural Transm. 2019, 126, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hulsmann, J.; Riederer, P. The nigral coup in Parkinson’s disease by α-synuclein and its associated rebels. Cells 2021, 10, 598. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Llerena, C.; Phillips, A.; Garcia-Reitboeck, P.; Hardy, J.; Pocock, J.M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 2016, 36, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 235–263. [Google Scholar] [CrossRef]
- Spargo, E.; Everall, I.P.; Lantos, P.L. Neuronal loss in the hippocampus in Huntington’s disease: A comparison with HIV infection. J. Neurol. Neurosurg. Psychiatry 1993, 56, 487–491. [Google Scholar] [CrossRef]
- Giralt, A.; Saavedra, A.; Alberch, J.; Pérez-Navarro, E. Cognitive dysfunction in Huntington’s disease: Humans, mouse models and molecular mechanisms. J. Huntingt. Dis. 2012, 1, 155–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular infiltration in traumatic brain injury. J. Neuroinflamm. 2020, 17, 328. [Google Scholar] [CrossRef]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef] [Green Version]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T cells get ready to tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Sevenich, L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front. Immunol. 2018, 9, 697. [Google Scholar] [CrossRef] [PubMed]
- Rueda-Carrasco, J.; Hong, S. The jekyll and hyde of TREM2. Trends Neurosci. 2020, 43, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Augusto-Oliveira, M.; Arrifano, G.P.; Lopes-Araújo, A.; Santos-Sacramento, L.; Takeda, P.Y.; Anthony, D.C.; Malva, J.O.; Crespo-Lopez, M.E. What do microglia really do in healthy adult brain? Cells 2019, 8, 1293. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Leak, R.K.; Hu, X. Neurotransmitter receptors on microglia. Stroke Vasc. Neurol. 2016, 1, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Ann. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Chagas, L.; Sandre, P.C.; Ribeiro, E.; Ribeiro, N.; Marcondes, H.; Oliveira Silva, P. environmental signals on microglial function during brain development, neuroplasticity, and disease. Int. J. Mol. Sci. 2020, 21, 2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Robinton, D.; Stevens, B. Microglia and the brain: Complementary partners in development and disease. Ann. Rev. Cell Dev. Biol. 2018, 34, 523–544. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Med. Inflamm. 2013, 480739. [Google Scholar] [CrossRef] [Green Version]
- Lecours, C.; Bordeleau, M.; Cantin, L.; Parent, M.; Di Paolo, T.; Tremblay, M.E. Microglial implication in Parkinson’s disease: Loss of beneficial physiological roles or gain of inflammatory functions? Front. Cell Neurosci. 2018, 12, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.S.Y.; Chao, Y.X.; Rötzschke, O.; Tan, E.K. New insights into immune-mediated mechanisms in Parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 9302. [Google Scholar] [CrossRef]
- Bartels, A.L.; Leenders, K.L. Cyclooxygenase and neuroinflammation in Parkinsons disease neurodegeneration. Curr. Neuropharmacol. 2010, 8, 62. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiology 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Artaud, F.; Cormier-Dequaire, F.; Rascol, O.; Durif, F.; Derkinderen, P. Examining the reserve hypothesis in Parkinson’s disease: A longitudinal study. Mov. Disord. 2019, 34, 1663–1671. [Google Scholar] [CrossRef]
- Pajares, M.I.; Rojo, A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s disease: Mechanisms and therapeutic implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef] [PubMed]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation—An Interesting interplay in Parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Kouli, A.; Camacho, M.; Allinson, K.; Williams-Gray, C.H. Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol. Commun. 2020, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.V. Toll signaling pathways in the innate immune response. Curr. Opin. Immunol. 2000, 12, 13–19. [Google Scholar] [CrossRef]
- May, M.J.; Ghosh, S. Signal transduction through NF-kappa B. Immunol. Today 1998, 19, 80–88. [Google Scholar] [CrossRef]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanke, M.L.; Kielian, T. Toll-like receptors in health and disease in the brain: Mechanisms and therapeutic potential. Clin. Sci. 2011, 121, 367–387. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Piccinini, A.M.; Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotta, T.; Porro, C.; Calvello, R.; Panaro, M.A. Biological role of toll-like receptor-4 in the brain. J. Neuroimmunol. 2014, 268, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Molteni, M.; Gemma, S.; Rossetti, C. The role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, C.D.; Choi, M.L.; Ryten, M.; Hopkins, L.; Drews, A.; Botía, J.A.; Iljina, M.; Rodrigues, M.; Gagliano, S.A.; Gandhi, S.; et al. Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol. 2019, 137, 103–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, W.H.; Jeon, M.T.; Leem, E.; Won, S.Y.; Jeong, K.H.; Park, S.J.; McLean, C.; Lee, S.J.; Jin, B.K.; Jung, U.J.; et al. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: A potential pathogenic mechanism in Parkinson’s disease. Sci. Rep. 2015, 5, 14764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.A. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef] [Green Version]
- Panaro, M.A.; Lofrumento, D.D.; Saponaro, C.; De Nuccio, F.; Cianciulli, A.; Mitolo, V.; Nicolardi, G. Expression of TLR4 and CD14 in the central nervous system (CNS) in a MPTP mouse model of Parkinson’s-like disease. Immunopharmacol. Immunotoxicol. 2008, 30, 729–740. [Google Scholar] [CrossRef]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Han, X.; Xue, L.; Zhu, K.; Liu, H.; Xie, A. Association of TLR4 gene polymorphisms with sporadic Parkinson’s disease in a Han Chinese population. Neurol. Sci. 2015, 36, 1659–1665. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [Green Version]
- Viceconte, N.; Burguillos, M.A.; Herrera, A.J.; De Pablos, R.M.; Joseph, B.; Venero, J.L. Neuromelanin activates proinflammatory microglia through a caspase-8-dependent mechanism. J. Neuroinflamm. 2015, 12, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouli, A.; Horne, C.B.; Williams-Gray, C.H. Toll-like receptors and their therapeutic potential in Parkinson’s disease and α-synucleinopathies. Brain Behav. Immun. 2019, 81, 41–51. [Google Scholar] [CrossRef]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef] [Green Version]
- Leitner, G.R.; Wenzel, T.J.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targ. 2019, 23, 865–882. [Google Scholar] [CrossRef]
- Campolo, C.; Paterniti, I.; Siracusa, R.; Filippone, A.; Esposito, E.; Cuzzocrea, S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav. Immun. 2019, 76, 236–247. [Google Scholar] [CrossRef]
- Loiarro, M.; Ruggiero, V.; Sette, C. Targeting TLR/IL-1R signalling in human diseases. Mediat. Inflamm. 2010, 674363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, G.M.; Medzhitov, R. Toll-like receptor signaling pathways. Science 2003, 300, 1524–1525. [Google Scholar] [CrossRef]
- Rajaiah, R.; Perkins, D.J.; Ireland, D.D.C.; Vogel, S.N. CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, 8391–8396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Deng, S.L.; Lian, Z.X.; Yu, K. Roles of toll-like receptors in nitroxidative stress in mammals. Cells 2019, 8, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Targ. Ther. 2020, 5. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. The inflammasome in times of COVID-19. Front. Immunol. 2020, 11, 583373. [Google Scholar] [CrossRef]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Rezaei, N. Role of Toll-like receptors in the pathogenesis of COVID-19. J. Med. Virol. 2021, 93, 2735–2739. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R. Innate receptor activation patterns Involving TLR and NLR synergisms in COVID-19, ALI/ARDS and sepsis cytokine storms: A review and model making novel predictions and therapeutic suggestions. Int. J. Mol. Sci. 2021, 22, 2108. [Google Scholar] [CrossRef] [PubMed]
- Khanolkar, A.; Hartwig, S.M.; Haag, B.A.; Meyerholz, D.K.; Harty, J.T.; Varga, S.M. Toll-like receptor 4 deficiency increases disease and mortality after mouse hepatitis virus type 1 infection of susceptible C3H mice. J. Virol. 2009, 83, 8946–8956. [Google Scholar] [CrossRef] [Green Version]
- Petruk, G.; Puthia, M.; Petrlova, J.; Samsudin, F.; Strömdahl, A.C.; Cerps, S. SARS-CoV-2 Spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J. Mol. Cell. Biol. 2021, 12, 916–932. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sharma, A.R.; Mallick, B.; Sharma, G.; Lee, S.S.; Chakraborty, C. Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex. Infect. Genet. Evol. 2020, 85, 104587. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Jin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and toll-like receptor 4 (TLR4): SARS-CoV-2 may bind and activate TLR4 to Increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediat. Inflamm. 2021. [Google Scholar] [CrossRef] [PubMed]
- Brandão, S.C.S.; Ramos, J.O.X.; Dompieri, L.T.; Godoi, E.T.A.M.; Figueiredo, J.L.; Sarinho, E.S.C.; Chelvanambi, S.; Aikawa, M. Is toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine Growth Factor Rev. 2020, 58, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.M.; Lee, S.G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 patients upregulate toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. J. Korean Med. Sci. 2020, 35, e343. [Google Scholar] [CrossRef] [PubMed]
- Anderson, F.L.; von Herrmann, K.M.; Andrew, A.S.; Kuras, Y.I.; Young, A.L.; Scherzer, C.R.; Hickey, W.F.; Lee, S.L.; Havrda, M.C. Plasma-borne indicators of inflammasome activity in Parkinson’s disease patients. NPJ Parkinsons Dis. 2021, 7. [Google Scholar] [CrossRef]
- Lucas, K.; Maes, M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: Possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef]
- Beraud, D.; Twomey, M.; Bloom, B.; Mittereder, A.; Neitzke, K.; Ton, V.; Chasovskikh, S.; Mhyre, T.; Maguire-Zeiss, K. α-synuclein alters toll-like receptor expression. Front. Naurosci. 2011, 5, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, L.C.; Finkelstein, D.I.; Bush, A.I.; Evans, A.H.; Barnham, K.J. Parkinsonism as a third wave of the COVID-19 pandemic? J. Parkinsons Dis. 2020, 10, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Ter Meulen, V. Coronaviruses: A challenge of today and a call for extended human postmortem brain analyses. J. Neural Transm. 2020, 127, 1217–1228. [Google Scholar] [CrossRef]
- Matsuda, K.; Park, C.H.; Sunden, Y.; Kimura, T.; Ochiai, K.; Kida, H.; Umemura, T. The vagus nerve is one route of transneural invasion for intranasally inoculated influenza a virus in mice. Vet. Pathol. 2004, 41, 101–107. [Google Scholar] [CrossRef]
- Chasey, D.; Alexander, D.J. Morphogenesis of avian infectious bronchitis virus in primary chick kidney cells. Arch. Virol. 1976, 52, 101–111. [Google Scholar] [CrossRef]
- Guo, Y.; Korteweg, C.; McNutt, M.A.; Gu, J. Pathogenetic mechanisms of severe acute respiratory syndrome. Virus Res. 2008, 133, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Pesce, M.; Seguella, L.; Sanseverino, W.; Lu, J.; Sarnelli, G. Can the enteric nervous system be an alternative entrance door in SARS-CoV2 neuroinvasion? Brain Behav. Immun. 2020, 87, 93–94. [Google Scholar] [CrossRef]
- Beyrouti, R.; Adams, M.E.; Benjamin, L.; Cohen, H.; Farmer, S.F.; Goh, Y.Y.; Humphries, F.; Jäger, H.R.; Losseff, N.A.; Perry, R.J.; et al. Characteristics of ischaemic stroke associated with COVID-19. J. Neurol. Neurosurg. Psychiatry 2020, 91, 889–891. [Google Scholar] [CrossRef]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front. Neurol. 2021, 11, 573095. [Google Scholar] [CrossRef]
- Jones, A.R.; Overly, C.C.; Sunkin, S.M. The allen brain atlas: 5 years and beyond. Nat. Rev. Neurosci. 2019, 10, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Merello, M.; Bhatia, K.P.; Obeso, J.A. SARS-CoV-2 and the risk of Parkinson’s disease: Facts and fantasy. Lancet Neurol. 2021, 20, 94–95. [Google Scholar] [CrossRef]
- Erickson, M.A.; Rhea, E.M.; Knopp, R.C.; Banks, W.A. Interactions of SARS-CoV-2 with the Blood–Brain barrier. Int. J. Mol. Sci. 2021, 22, 2681. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Vieites Prado, A.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef]
- Desforges, M.; Le Coupanec, A.; Brison, E.; Meessen-Pinard, M.; Talbot, P.J. Neuroinvasive and neurotropic human respiratory coronaviruses: Potential neurovirulent agents in humans. Adv. Exp. Med. Biol. 2014, 807, 75–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bale, J.F., Jr. Virus and immune-mediated encephalitides: Epidemiology, diagnosis, treatment, and prevention. Pediatr. Neurol. 2015, 53, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailles, A.; Stahl, J.P.; Bloch, K.C. Update and new insights in encephalitis. Clin. Microbiol. Infect. 2017, 23, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Bohmwald, K.; Gálvez, N.M.S.; Ríos, M.; Kalergis, A.M. Neurologic alterations due to respiratory virus infections. Front. Cell Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Jiangm, D.; Huangm, J.T. SARS-CoV-2 detected in cerebrospinal fluid by PCR in a case of COVID-19 encephalitis. Brain Behav. Immun. 2020, 87, 149. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Mahajan, S.D. SARS-COV2 alters blood brain barrier integrity contributing to neuro-inflammation. J. Neuroimmune Pharmacol. 2021, 16, 4–6. [Google Scholar] [CrossRef]
- Alquisiras-Burgos, I.; Peralta-Arrieta, I.; Alonso-Palomares, L.A. Neurological complications associated with the blood-brain barrier damage induced by the inflammatory response during SARS-CoV-2 infection. Mol. Neurobiol. 2020, 58, 1–16. [Google Scholar] [CrossRef]
- Kustin, T.; Ling, G.; Sharabi, S.; Ram, D.; Friedman, N.; Zuckerman, N.; Bucris, E.D.; Glatman-Freedman, A.; Stern, A.; Mandelboim, M. A method to identify respiratory virus infections in clinical samples using next-generation sequencing. Sci. Rep. 2019, 9, 2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzini, A.; Padovani, A. Lifting the mask on neurological manifestations of COVID-19. Nat. Rev. Neurol. 2020, 16, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Guerrero, A.; Laespada-García, M.I.; Gómez-Grande, A.; Ruiz-Ortiz, M.; Blanco-Palmero, V.A.; Azcarate-Diaz, F.J.; Rábano-Suárez, P.; Álvarez-Torres, E. Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 2020, 95, e2109–e2118. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.A. Case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Faber, I.; Brandão, P.R.; Menegatti, F.; de Carvalho Bispo, D.D.; Maluf, F.B.; Cardoso, F. Coronavirus disease 2019 and Parkinsonism: A non-post-encephalitic case. Mov. Disord. 2020, 35, 1721–1722. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.E.; Ho, A.P.; Ho, M.A.; Ho, E.C. Case report of familial COVID-19 cluster associated with high prevalence of anosmia, ageusia, and gastrointestinal symptoms. IDCases 2020, 22, e00975. [Google Scholar] [CrossRef] [PubMed]
- Tarakad, A.; Jankovic, J. Anosmia and ageusia in Parkinson’s disease. Int. Rev. Neurobiol. 2017, 133, 541–556. [Google Scholar] [CrossRef]
- Sepehrinezhad, A.; Shahbazi, A.; Negah, S.S. COVID-19 virus may have neuroinvasive potential and cause neurological complications: A perspective review. J. Neurovirol. 2020, 26, 324–329. [Google Scholar] [CrossRef]
- Espinosa, P.S.; Rizvi, Z.; Sharma, P.; Hindi, F.; Filatov, A. Neurological complications of Coronavirus Disease (COVID-19): Encephalopathy, MRI brain and cerebrospinal fluid findings: Case 2. Cureus 2020, 12, e7930. [Google Scholar] [CrossRef]
- Zhang, J.; Hao, Y.; Ou, W.; Ming, F.; Liang, G.; Cai, Q.; Dong, S.; Hu, S.; Wang, W.; Wei, S. Serum interleukin-6 is an indicator for severity in 901 patients with SARS-CoV-2 infection: A cohort study. J. Transl. Med. 2020, 18, 406–413. [Google Scholar] [CrossRef]
- Pereira, J.R.; Santos, L.V.D.; Santos, R.M.S.; Campos, A.L.F.; Pimenta, A.L.; de Oliveira, M.S. IL-6 serum levels are elevated in Parkinson’s disease patients with fatigue compared to patients without fatigue. J. Neurol. Sci. 2016, 370, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Veselý, B.; Dufek, M.; Thon, V.; Brozman, M.; Királová, S.; Halászová, T.; Koritáková, E.; Rektor, I. Interleukin 6 and complement serum level study in Parkinson’s disease. J. Neural Transm. 2018, 125, 875–881. [Google Scholar] [CrossRef]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E.; Beckham, J.D. Alpha-synuclein expression restricts RNA viral infections in the brain. J. Virol. 2015, 90, 2767–2782. [Google Scholar] [CrossRef] [Green Version]
- Ait Wahmane, S.; Achbani, A.; Ouhaz, Z.; Elatiqi, M.; Belmouden, A.; Nejmeddine, M. The possible protective role of α-synuclein against severe acute respiratory syndrome Coronavirus 2 infections in patients with Parkinson’s disease. Mov. Disord. 2020, 35, 1293–1294. [Google Scholar] [CrossRef]
- Semerdzhiev, S.A.; Fakhree, M.A.A.; Segers-Nolten, I.; Blum, C.; Claessens, M.M.A.E. Interactions between SARS-CoV-2 N-protein and α-synuclein accelerate amyloid formation. BioRxiv 2021. [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Marreiros, R.; Müller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hänsch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef]
- Chaná-Cuevas, P.; Salles-Gándara, P.; Rojas-Fernandez, A.; Salinas-Rebolledo, C.; Milán-Solé, A. The potential role of SARS-COV-2 in the pathogenesis of Parkinson’s disease. Front. Neurol. 2020, 11, 1044. [Google Scholar] [CrossRef] [PubMed]
- Tulisiak, C.T.; Mercado, G.; Peelaerts, W.; Brundin, L.; Brundin, P. Can infections trigger alpha-synucleinopathies? Prog. Mol. Biol. Transl. Sci. 2019, 168, 299–322. [Google Scholar] [CrossRef]
- Labrie, V.; Brundin, P. Alpha-synuclein to the rescue: Immune cell recruitment by alpha-synuclein during gastrointestinal infection. J. Innate Immun. 2017, 9, 80. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematical representation of TLR4 activation in microglia and consequent dopaminergic neuronal damage in the substantia nigra. A number of pro-inflammatory stimuli can promote the activation of the intracellular TLR4 signaling pathway in microglia. Upon infection or injury, DAMPs or PAMPs released in the extracellular milieu via exocytosis from neighboring neurons are sensed by microglial TLR4 for degradation and clearance. If compromised or if prolonged, this process can lead to further inflammatory signaling in which the engagement of myeloid differentiating primary response gene 88 (MyD88) or TIR-domain containing adapter inducing interferon β (TRIF) provokes the activation of downstream signaling cascades that lead to the proteasomal degradation of kB inhibitors (IkB), release, and nuclear translocation of the NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells) transcription factor. The transcriptional activation of specific genes induces the release of proinflammatory cytokines, that in turn can cause neuronal damage, pathological modification of α-SYN (monomers, oligomers, and fibrils, truncated and phosphorylated forms), and aggregation in Lewy and neurites bodies—the pathological hallmarks of degenerating neurons in Parkinson’s disease. Endosomal TLRs sense bacterial or viral nucleic acids.
Figure 1.
Schematical representation of TLR4 activation in microglia and consequent dopaminergic neuronal damage in the substantia nigra. A number of pro-inflammatory stimuli can promote the activation of the intracellular TLR4 signaling pathway in microglia. Upon infection or injury, DAMPs or PAMPs released in the extracellular milieu via exocytosis from neighboring neurons are sensed by microglial TLR4 for degradation and clearance. If compromised or if prolonged, this process can lead to further inflammatory signaling in which the engagement of myeloid differentiating primary response gene 88 (MyD88) or TIR-domain containing adapter inducing interferon β (TRIF) provokes the activation of downstream signaling cascades that lead to the proteasomal degradation of kB inhibitors (IkB), release, and nuclear translocation of the NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells) transcription factor. The transcriptional activation of specific genes induces the release of proinflammatory cytokines, that in turn can cause neuronal damage, pathological modification of α-SYN (monomers, oligomers, and fibrils, truncated and phosphorylated forms), and aggregation in Lewy and neurites bodies—the pathological hallmarks of degenerating neurons in Parkinson’s disease. Endosomal TLRs sense bacterial or viral nucleic acids.

Figure 2.
Toll-like receptor 4 (TLR4) signaling cascade resulting from the interaction between TLR4 and SARS-CoV-2 S protein. (a). Schematic diagram of the primary structural proteins of SARS-CoV-2.(b). The interaction between TLR4 and the SARS-CoV-2 S protein can trigger an intracellular TLR4 signaling cascade that can be one of the factors leading to the cytokine storm and neuroinflammation in severe COVID-19 patients. SARS-CoV-2: Severe Acute Respiratory Syndrome Coronavirus 2; Coronavirus disease. CD14: cluster of differentiation 14; MD2: myeloid differential protein-2; MyD88: myeloid differentiating primary response gene 88; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; IRAK: interleukin-1 receptor-associated kinases; TRIF: TIR-domain containing adapter inducing interferon β; TRAM: TRIF-related adaptor molecule; TRAF: tumor necrosis factor receptor-associated factor; IFN: interferon.
Figure 2.
Toll-like receptor 4 (TLR4) signaling cascade resulting from the interaction between TLR4 and SARS-CoV-2 S protein. (a). Schematic diagram of the primary structural proteins of SARS-CoV-2.(b). The interaction between TLR4 and the SARS-CoV-2 S protein can trigger an intracellular TLR4 signaling cascade that can be one of the factors leading to the cytokine storm and neuroinflammation in severe COVID-19 patients. SARS-CoV-2: Severe Acute Respiratory Syndrome Coronavirus 2; Coronavirus disease. CD14: cluster of differentiation 14; MD2: myeloid differential protein-2; MyD88: myeloid differentiating primary response gene 88; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; IRAK: interleukin-1 receptor-associated kinases; TRIF: TIR-domain containing adapter inducing interferon β; TRAM: TRIF-related adaptor molecule; TRAF: tumor necrosis factor receptor-associated factor; IFN: interferon.

Figure 3.
SARS-CoV-2 neuroinvasion and neuropathogenesis. SARS-CoV-2 may cause the disruption of the BBB integrity to gain access to the CNS. Viral invasion initiates inflammatory signaling involving the activation of microglia and release of abundant levels of proinflammatory cytokines/chemokines, which in turn can disrupt the BBB and increase its permeability. The inflammatory response can cause the neuronal accumulation of α-SYN and neuronal damage. The aggregates of α-SYN released from the neurons can induce microglia activation and initiate a vicious circle. IL: interleukin; TNFα: tumour necrosis factor alpha, IFN-γ: Interferon-gamma; Cox-2; cyclooxygenase-2; NOS: nitric oxide synthase; ROS: reactive oxygen species.
Figure 3.
SARS-CoV-2 neuroinvasion and neuropathogenesis. SARS-CoV-2 may cause the disruption of the BBB integrity to gain access to the CNS. Viral invasion initiates inflammatory signaling involving the activation of microglia and release of abundant levels of proinflammatory cytokines/chemokines, which in turn can disrupt the BBB and increase its permeability. The inflammatory response can cause the neuronal accumulation of α-SYN and neuronal damage. The aggregates of α-SYN released from the neurons can induce microglia activation and initiate a vicious circle. IL: interleukin; TNFα: tumour necrosis factor alpha, IFN-γ: Interferon-gamma; Cox-2; cyclooxygenase-2; NOS: nitric oxide synthase; ROS: reactive oxygen species.

Figure 4.
Possible direct interaction between the spike protein and α-sinuclein. (a) SARS-CoV-2 genome organization. Open Reading Frames (ORFs); Envelope (E); Nucleocapside (N); Membrane protein (M). (b) Schematic depiction of α-synuclein structure. N-terminus, NAC (non-amyloid-β component) region, and C-terminus are coloured blue, pink and red, respectively. (NAC). (c) The possible direct interaction between SARS-CoV-2 and α-SYN could start a conformational shift of the monomeric protein and accelerate the formation of toxic multimeric protein species, such as oligomers/protofibrils and fibril aggregates, resulting in neuronal death.
Figure 4.
Possible direct interaction between the spike protein and α-sinuclein. (a) SARS-CoV-2 genome organization. Open Reading Frames (ORFs); Envelope (E); Nucleocapside (N); Membrane protein (M). (b) Schematic depiction of α-synuclein structure. N-terminus, NAC (non-amyloid-β component) region, and C-terminus are coloured blue, pink and red, respectively. (NAC). (c) The possible direct interaction between SARS-CoV-2 and α-SYN could start a conformational shift of the monomeric protein and accelerate the formation of toxic multimeric protein species, such as oligomers/protofibrils and fibril aggregates, resulting in neuronal death.

Figure 5.
Possible mechanism proposed for α-synuclein aggregation in dopaminergic neurons. The binding of the S protein to the ACE2 receptor mediates the virus entry into dopaminergic cells. In the host cells, the virus can induce endoplasmic reticulum (ER) stress and adaptive unfolded protein response (UPR) and ubiquitin proteasome system (UPS) activation, leading to impairment of proteostasis, α-SYN aggregation, and neurodegeneration. A similar mechanism can be triggered by SARS-CoV-2-induced TLR4 activation.
Figure 5.
Possible mechanism proposed for α-synuclein aggregation in dopaminergic neurons. The binding of the S protein to the ACE2 receptor mediates the virus entry into dopaminergic cells. In the host cells, the virus can induce endoplasmic reticulum (ER) stress and adaptive unfolded protein response (UPR) and ubiquitin proteasome system (UPS) activation, leading to impairment of proteostasis, α-SYN aggregation, and neurodegeneration. A similar mechanism can be triggered by SARS-CoV-2-induced TLR4 activation.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Conte, C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. Int. J. Mol. Sci. 2021, 22, 7135. https://doi.org/10.3390/ijms22137135
AMA Style
Conte C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. International Journal of Molecular Sciences. 2021; 22(13):7135. https://doi.org/10.3390/ijms22137135
Chicago/Turabian StyleConte, Carmela. 2021. "Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4" International Journal of Molecular Sciences 22, no. 13: 7135. https://doi.org/10.3390/ijms22137135
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.