
Abstract
Porcine reproductive and respiratory syndrome (PRRS) is one of the most economically devastating infectious diseases in the global swine industry. A rapid and sensitive on-site detection method for PRRS virus (PRRSV) is critically important for diagnosing PRRS. In this study, we established a method that combines reverse transcription recombinase polymerase amplification (RT-RPA) with a lateral flow dipstick (LFD) for detecting North American PRRSV (PRRSV-2). The primers and probe were designed based on the conserved region of all complete PRRSV-2 genomic sequences available in China (n = 512) from 1996 to 2020. The detection limit of the assay was 5.6 × 10-1 median tissue culture infection dose (TCID50) per reaction within 30 min at 42 °C, which was more sensitive than that of reverse transcription polymerase chain reaction (RT-PCR) (5.6 TCID50 per reaction). The assay was highly specific for the epidemic lineages of PRRSV-2 in China and did not cross-react with pseudorabies virus, porcine circovirus 2, classical swine fever virus, or porcine epidemic diarrhea virus. The assay performance was evaluated by testing 179 samples and comparing the results with those of quantitative RT-PCR (RT-qPCR). The results showed that the detection coincidence rate of RT-RPA and RT-qPCR was 100% when the cycle threshold values of RT-qPCR were < 32. The assay provides a new alternative for simple and reliable detection of PRRSV-2 and has great potential for application in the field.
Similar content being viewed by others

Introduction
Porcine reproductive and respiratory syndrome (PRRS) is an economically disastrous infectious disease for the global swine industry [1, 2]. The disease was first reported in 1987 in America and became widespread in Europe and Asia in subsequent years. It causes annual losses of 500–600 million dollars in the US swine industry, and even more in Asia, due to reproductive disorders in pregnant sows and respiratory symptoms in pigs [3, 4]. PRRS virus (PRRSV), the causative agent, is a single-stranded positive-sense RNA virus belonging to the family Arteriviridae of the order Nidovirales [5]. It can be classified into two genotypes, namely the European type (PPRSV-1) and the North American type (PRRSV-2) [6]. Based on the genetic distance between ORF5 sequences, PRRSV-2 is divided into nine lineages (L1–L9), and the main epidemic lineages in China are L1, L3, L5, and L8 [7,8,9]. PRRSV was first isolated in China in 1996 [10]. In 2006, highly pathogenic PRRSV (HP-PRRSV) emerged in Jiangxi Province and then spread rapidly to most of the other provinces of China, causing high fever and a high rate of mortality in pigs of all ages [11, 12]. In 2012, a new subgroup of PRRSV strains was identified and was named NADC30-like, due to its similarity to the American strain NADC30. The strains in the NADC30-like subgroup have increased rapidly from 2015 to 2017 [13, 14]. PRRSV has a high level of diversity due to its high variability and its tendency to undergo recombination, which renders the prevention and control of PRRS more challenging.
Quick and reliable detection methods are of great significance for timely diagnosis, control, and prevention of PRRS. Various detection methods targeting PRRSV nucleic acids, antigens, and antibodies are currently available. Among these approaches, enzyme-linked immunosorbent assay [15, 16], polymerase chain reaction (PCR) [17, 18], and indirect immunofluorescence assay [19] are the most widely applied. However, these technologies have limitations when applied in the field because they require professional equipment and technicians and are time-consuming. Therefore, it is necessary to develop a speedy, sensitive, and equipment-free diagnostic assay for detecting PRRSV. Recombinase polymerase amplification (RPA), which was first reported in 2006, is an isothermal amplification technique [20] that does not require thermal denaturation of the template and can be operated at a low and constant temperature in a simple water bath, or even at body temperature. To date, RPA-based detection has been successfully applied for the detection of severe acute respiratory syndrome coronavirus 2, classical swine fever virus (CSFV), African swine fever virus, porcine circovirus 2 (PCV2), foot-and-mouth disease virus, porcine deltacoronavirus, and other viruses [21,22,23,24,25,26].
In this study, a reverse transcription RPA (RT-RPA) assay targeting the conserved region of the ORF6 gene was developed for rapid visual detection of PRRSV-2. The amplicons were detected in 3 min, using a lateral flow dipstick (LFD), and the assay exhibited better sensitivity than RT-PCR. We also compared the performance of RT-RPA in detecting clinical samples using a published RT-qPCR assay.
Materials and methods
Viruses and cell culture isolates
HP-PRRSV strain HuN4 (GenBank accession no. EF635006), the NADC30-like PRRSV strain HeB108 (GenBank accession no. MN046224), CSFV, PCV2, pseudorabies virus (PRV), and porcine epidemic diarrhea virus (PEDV) were preserved in our laboratory. Forty-four PRRSV-2 cell culture isolates from samples from Shandong and Jiangsu provinces were preserved in our laboratory. MARC-145 cells were cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO, USA) with 10% fetal bovine serum (ExCell, Australia) at 37 °C in a 5% CO2 atmosphere [27].
Clinical samples
A total of 135 field samples, including samples from lung (71), spleen (17), kidney (6), intestine (5), lymph nodes (5), and liver (3) as well as 8 throat swabs, and 20 blood samples were collected from different farms in Guangdong, Hebei, Shandong, Zhejiang, Heilongjiang, and Liaoning provinces of China from 2018 to 2019.
Nucleic acid extraction
Viral RNA of CSFV, PEDV, and PRRSV-2 of different lineages (PRRSV-L1, PRRSV-L3, PRRSV-L5, and PRRSV-L8) was extracted using a QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer's instructions. The extracted RNA was eluted with 50 μL of nuclease-free water and stored at -80 ℃. Viral DNA of PRV and PCV2 was extracted using a TIANamp Virus DNA/RNA Kit (TIANGEN, Beijing, China) according to the manufacturer's instructions. The extracted DNA was eluted with 50 μL of nuclease-free water and stored at -20 °C until use.
Primer and probe design for RT-RPA
All complete genomic sequences of PRRSV-2 from China (n = 512) that were available in the GenBank database by July 2020 were aligned using DNASTAR (DNASTAR Inc., Madison, WI, USA) to identify the most conserved region. RPA primers and a probe were designed based on the conserved region of the ORF6 gene and were as follows: RPA sense primer, 5ʹ- GGCCGCAAGTACATTCTGGCCCCTGCCCACCACG-3ʹ; RPA antisense primer, 5ʹ-Biotin-GGCCGCAAGTACATTCTGGCCCCTGCCCACCACG-3ʹ; RPA probe primer, 5ʹ-FAM-AACGGCAAGTGATAACCACGCATTTGTCG[THF]CCGGCGTCCCGGCTC-C3spacer-3ʹ. The primers and probe were synthesized by Comate Bioscience Co., Ltd. (Jilin, China).
Development of the PRRSV-2 RT-RPA assay with LFD
The RPA assays were conducted using an RT-RPA nfo kit (GenDx, Utrecht, The Netherlands) according to the manufacturer’s instructions. The reaction mixtures consisted of 20 μL of rehydration buffer, 2 μL of 350 mM magnesium acetate, 2.1 μL of each primer pair (10 μM), 0.6 μL of probe (10 μM), 0.5 μL of RNase inhibitor, 20.7 μL of nuclease-free water, and 2 μL of viral RNA or DNA. All reagents except the template and magnesium acetate were prepared in a master mix, which was dispensed into 0.2-mL reaction tubes containing the fluorescence detection reagent (freeze-dried reaction pellets). Briefly, 2 μL of magnesium acetate was slowly pipetted into the tube lid, and 2 μL of template was added to the tubes. The reaction tubes were immediately placed in a water bath to initiate amplification. The amplified product was diluted 100 times with sample diluent, and the lateral flow dipstick (Zoonbio Biotechnology, China) was dipped into 80 μL of the diluted product for 2–3 min.
Real-time qPCR assay
An RT-qPCR assay targeting the ORF6 gene of PRRSV, developed by Chen et al., was carried out using Quant Studio 5 (Applied Biosystems, Waltham, MA, USA) according to the manufacturer’s instructions with some modifications [28]. The 25-μL reaction mixture contained 12.5 μL of 2 × Premix Ex Taq, 1 μL of probe (10 μM), 0.5 μL of each pair of primers (10 μM), 2 μL of RNA, and 8.5 μL of ddH2O. The amplification conditions were 95 ℃ for 30 s, followed by 30 cycles at 95 ℃ for 5 s and 60 ℃ for 30 s. All experiments were performed with at least three replicates.
Sensitivity of the RT-RPA assay
Viral RNA was extracted from the PRRSV-2 strain at a concentration of 106 median tissue culture infection dose (TCID50)/mL, and the final RNA concentration was equivalent to 2.8 × 103 TCID50/μL. Through tenfold gradient dilution, different RNA concentrations, ranging from 2.8 × 103 TCID50 to 2.8 × 10-3 TCID50, were obtained. Briefly, 2 µL of each dilution was used to evaluate the sensitivity of the assay, and the amplicons were detected simultaneously by LFD and agarose gel electrophoresis.
Specificity of the RT-RPA assay
To evaluate the specificity of the RT-RPA for PRRSV-2, different lineages of PRRSV-2 (PRRSV-L1, PRRSV-L3, PRRSV-L5, and PRRSV-L8) as well as selected swine pathogens, including CSFV, PCV-2, PRV, and PEDV, were tested.
Statistical analysis
Kappa analysis was carried out using SPSS 22.0 (IBM, Armonk, NY, USA) to compare the detection results obtained with the RT-RPA and the RT-qPCR assays and to determine their correlation. A p-value of <0.05 was considered statistically significant.
Results
Establishment and optimization of the reaction parameters of the PRRSV-2 RT-RPA assay
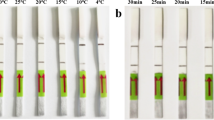
Different reaction temperatures (20 °C, 23 °C, 25 °C, 27 °C, 30 °C, 33 °C, 35 °C, 37 °C, 39 °C, 42 °C, 45 °C, 48 °C, 50 °C, 53 °C, and 55 °C) were tested, and the products were detected by agarose gel electrophoresis. As shown in Fig. 1a, no amplification products were observed in reactions incubated at <30 °C or ≥48 °C. Weak bands were observed at 30 °C, and there was no difference in the amount of amplification at 42 °C and 45 °C. Therefore, 42 °C was selected as the optimal reaction temperature. The reaction mixture was incubated at 42 °C for different lengths of time to determine the optimal incubation time. As shown in Fig. 1b, when the incubation time was < 5 min, no amplification products were observed, and only a faint band was observed after incubating for 10 min. There was no difference in the amount of amplification in the reactions incubated for 30 min and 35 min, and therefore, 30 min was selected as the optimal reaction time.

Optimization of the reaction temperature and time of the PRRSV-2 RT-RPA assay. A The assay functions in the temperature range of 30–45 ℃, with 42 ℃ or 45 ℃ being the optimal reaction temperature. B A positive signal is seen in 5 min, and the brightness of the band gradually increases with increasing amplification time. No difference was observed between 30 min and 35 min. M DNA marker
Sensitivity of the PRRSV-2 RT-RPA assay
For sensitivity analysis, tenfold serial dilutions of PRRSV-2 RNA ranging from 2.8 × 103 TCID50 to 2.8 × 10-3 TCID50 were performed. As shown in Fig. 2, the detection limit of PRRSV-2 RT-RPA was 5.6 × 10-1 TCID50 per reaction. However, when the amplicon was detected by agarose gel electrophoresis, the detection limit was only 5.6 TCID50 per reaction. Thus, detection of the RPA product of by LFD is more sensitive than detection by agarose gel electrophoresis.

Sensitivity of the PRRSV-2 RT-RPA assay. The amplification products of the PRRSV-2 RT-RPA were analyzed by LFD and agarose gel electrophoresis. A In the LFD, the sensitivity was 5.6 × 10-1 TCID50 per reaction. The top line on the LFD is the control line, and the bottom line is the test line. B In agarose gel electrophoresis, the sensitivity was 5.6 TCID50 per reaction. M DNA marker, NC negative control, LFD lateral flow dipstick
Specificity of the PRRSV-2 RT-RPA assay
The specificity of PRRSV-2 RT-RPA was evaluated using PRRSV-2 from different lineages (PRRSV-L1, PRRSV-L3, PRRSV-L5, and PRRSV-L8) and other swine pathogens, including CSFV, PCV2, PRV, and PEDV. As shown in Fig. 3, only PRRSV strains were detected, with no cross-reactions with other swine pathogens. The results indicate that the RT-RPA assay has good specificity for PRRSV-2.

Specificity of the PRRSV-2 RT-RPA assay. Four main lineages of PRRSV-2 in China (PRRSV-L1, PRRSV-L3, PRRSV-L5, and PRRSV-L8) and four other swine pathogens—CSFV, PCV2, PRV, and PEDV—were used as templates. NC negative control
Evaluation of the PRRSV-2 RT-RPA assay using viral isolates from cell culture
For preliminary evaluation of the detection rate and generality of the RT-RPA, 44 viral isolates of different lineages of PRRSV-2 from cell culture were simultaneously detected via RT-RPA and RT-qPCR. The results showed that all 44 samples were positive by both RT-qPCR and RT-RPA (Fig. 4 and Table 1).

Correlation between RT-RPA and RT-qPCR for detecting PRRSV-2. Each solid dot represents a sample. Red solid dots indicate that the sample tested positive by RT-qPCR, and green indicates a negative result.
Performance of the PRRSV-2 RT-RPA assay using clinical samples
A total of 135 clinical samples were tested using the RT-RPA assay, and the results were compared to those obtained using RT-qPCR. Out of 135 clinical samples, 84 tested positive by RT-RPA. The 84 clinical samples that tested positive by both RT-RPA and RT-qPCR were confirmed by sequencing to be true positives. However, 29 clinical samples with higher cycle threshold (Ct) values (32-35) tested positive by RT-qPCR but negative by RT-RPA (Fig. 4 and Table 1). The detection results for each sample obtained by RT-RPA and RT-qPCR are shown in Supplementary Table S1. Kappa analysis revealed that the kappa score between the RT-RPA and RT-qPCR methods was 0.52, indicating moderate consistency. In general, the detection coincidence rate of RT-RPA and RT-qPCR was 100% when the Ct values of RT-qPCR were higher than 35 or lower than 32, and the coincidence rate was only 15% when the Ct values were between 32 and 35 (Fig. 4 and Table 1).
Discussion
Since it was first identified in 1987, PRRS has been one of the acute viral diseases that typically leads to large economic losses to the global swine industry due to PRRSV-induced reproductive disorders in sows and respiratory diseases in growing pigs [29]. When an outbreak of PRRSV infection is not diagnosed in time, it cannot be treated in a timely manner, which increases the risk of transmission to other pigs [23], making it more difficult to prevent and control PRRS. Therefore, it is important to develop sensitive and convenient detection methods.
Although PCR has been widely used for detection of PRRSV with high sensitivity and specificity, its applicability is limited to laboratory detection, and it is difficult to use for on-site diagnosis. Isothermal nucleic acid amplification assays, including loop-mediated isothermal amplification assay (LAMP) [30], nucleic acid sequence-based amplification [31], rolling-circle amplification [32], and RPA [20] have been developed recently. These assays are quick and equipment-free and allow the reaction to be performed under isothermal conditions (such as in a water bath) without requiring thermal denaturation.
An RT-LAMP assay combined with metal indicators has been developed for the field detection of PRRSV; however, the reaction requires heat inactivation for 40 min at 58 °C, followed by 5 min at 80 °C, and also uses six primers, resulting in nonspecific amplification [33]. Compared to RT-LAMP, RPA requires a shorter time (5–30 min) and a lower operation temperature (25–42 ℃) [34]. Real-time RT-RPA for rapid detection of HP-PRRSV and classical PRRSV strains (C-PRRSV) have been reported [35]. Although these assays can rapidly and specifically detect PRRSV with a low detection limit, the detection methods are based on expensive scanning devices.
In this study, a novel PRRSV-2 RT-RPA assay combined with an LFD was established. To the best of our knowledge, this is the first report of a combination of RT-RPA and LFD to detect PRRSV-2. After the optimization of the reaction times and temperatures, the sensitivity of the assay was 5.6 × 10-1 TCID50 per reaction when performed at 42 °C for 30 min, exhibiting better sensitivity than RT-PCR. In addition, the assay was highly specific for the main lineages of PRRSV-2 in China and did not cross-react with PRV, PCV2, CSFV, or PEDV. This assay has several advantages. First, unlike the established PRRSV RT-RPA methods, it can detect not only HP-PRRSV and C-PRRSV but also NADC30-like strains, which are epidemic in many provinces of China, without relying on a scanning device. Second, the assay does not require thermal denaturation and can operate at a low and constant temperature, unlike RT-qPCR. Although RT-RPA has many advantages over RT-qPCR, it has some limitations when applied in the field. One potential limitation is that it is susceptible to contamination because the tube needs to be opened after amplification. Therefore, measures need to be taken to avoid contamination, such as opening and closing tubes carefully, changing gloves frequently, and separating the amplification area from the test area. Moreover, the sensitivity of the RT-RPA assay is lower than that of RT-qPCR and needs to be improved. As shown in Fig. 4, the RT-RPA assay fails to detect the virus in samples with a low viral load.
In summary, a visual nucleic acid detection method based on LFD was developed for PRRSV-2. This assay provides a new alternative for simple and reliable detection of PRRSV-2 and has great potential for application in the field and in resource-limited areas.
References
Wensvoort G, de Kluyver EP, Pol M (1992) Lelystad virus, the cause of porcine epidemic abortion and respiratory syndrome: a review of mystery swine disease research at Lelystad. Vet Microbiol 33:185–193
Neumann E, Kliebenstein J, Johnson C, Mabry J, Bush E, Seitzinger A, Green A, Zimmerman J, Nuemann EJ, Kleibenstein JB (2005) Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J Am Vet Med Assoc 227:385–392
Song J, Gao P, Kong C, Zhou L, Ge X, Guo X, Han J, Yang H (2019) The nsp2 hypervariable region of porcine reproductive and respiratory syndrome virus strain JXwn06 is associated with viral cellular tropism to primary porcine alveolar macrophages. J Virol 93:e01436-e1519
Zhou L, Yang H (2010) Porcine reproductive and respiratory syndrome in China. Virus Res 154:31–37
Kappes MA, Faaberg KS (2015) PRRSV structure, replication and recombination: origin of phenotype and genotype diversity. Virology 479–480:475–486
Benfield DA, Nelson E, Collins JE, Harris L, Goyal SM, Robison D, Christianson WT, Morrison RB, Gorcyca D (1992) Characterization of swine infertility and respiratory syndrome (SIRS) virus (isolate ATCC VR-2332). J Vet Diagn Investig 4:127–133
Shi M, Lam TT, Hon CC, Murtaugh MP, Davies PR, Hui RK, Li J, Wong LT, Yip CW, Jiang JW, Leung FC (2010) Phylogeny-based evolutionary, demographical, and geographical dissection of North American type 2 porcine reproductive and respiratory syndrome viruses. J Virol 84:8700–8711
Xu H, Song S, Zhao J, Leng C, Fu J, Li C, Tang YD, Xiang L, Peng J, Wang Q, Zhao H, An T, Cai X, Zhang H, Tian ZJ (2020) A potential endemic strain in China: NADC34-like porcine reproductive and respiratory syndrome virus. Transbound Emerg Dis 67:1730–1738
Zhao K, Ye C, Chang X-B, Jiang C-G, Wang S-J, Cai X-H, Tong G-Z, Tian Z-J, Shi M, An T-Q, Lyles DS (2015) Importation and recombination are responsible for the latest emergence of highly pathogenic porcine reproductive and respiratory syndrome virus in China. J Virol 89:10712–10716
Valícek L, Psikal I, Smíd B, Rodák L (1997) Isolation and identification of porcine reproductive and respiratory syndrome virus in cell cultures. Veterinární medicína 42:281–287
Tong GZ, Zhou YJ, Hao XF, Tian ZJ, An TQ, Qiu HJ (2007) Highly pathogenic porcine reproductive and respiratory syndrome, China. Emerg Infect Dis 13:1434–1436
Zhou YS, Yang X, Wang HN, Zhang AY, Zhang ZK, Kang RM, Zeng FY, Li HCJF (2012) Molecular characterization of a complete genome and 12 nsp2 genes of PRRSV of southwestern China. Food Environ Virol 4:102–114
Jiang Y, Li G, Yu L, Li L, Zhang Y, Zhou Y, Tong W, Liu C, Gao F, Tong G (2020) Genetic diversity of porcine reproductive and respiratory syndrome virus (PRRSV) from 1996 to 2017 in China. Front Microbiol 11:618
Lei Z, Wang Z, Ding Y, Ge X, Guo X, Yang H (2015) NADC30-like strain of porcine reproductive and respiratory syndrome virus, China. Emerg Infect Dis 21:2256–2257
Kittawornrat A, Prickett J, Wang C, Olsen C, Irwin C, Panyasing Y, Ballagi A, Rice A, Main R, Johnson J (2012) Detection of Porcine reproductive and respiratory syndrome virus (PRRSV) antibodies in oral fluid specimens using a commercial PRRSV serum antibody enzyme-linked immunosorbent assay. J Vet Diagn Invest 24(2):262–269
Xiao YH, Wang TT, Zhao Q, Wang CB, Lv JH, Nie L, Gao JM, Ma XC, Hsu WH, Zhou EMJT (2014) Development of indirect ELISAs for differential serodiagnosis of classical and highly pathogenic porcine reproductive and respiratory syndrome virus. Transbound Emerg Dis 61:341–349
Guo Z, Chen XX, Li X, Qiao S, Deng R, Zhang G (2019) Prevalence and genetic characteristics of porcine reproductive and respiratory syndrome virus in central China during 2016–2017: NADC30-like PRRSVs are predominant. Microb Pathog 135:103657
Peng Z, Zhao T, Liang W, Song W, Gao Z, Tang X, Chen H, Wu B (2017) RT-PCR detection of porcine reproductive and respiratory syndrome virus based on the ORF5 gene in mainland China, 2012–2015. Acta Virol 61:336–340
Wang FX, Yang Y, Liu X, He MH, Liu Y, Sun N, Zhu HW, Ren JQ, Wu H, Wen YJ (2017) Development of monoclonal antibody for differentiating porcine reproductive and respiratory syndrome virus and identification of a novel non-structural protein 2 epitope peptide. Virusdisease 28:408–415
Piepenburg O, Williams CH, Stemple DL, Armes NAJPB (2006) DNA detection using recombination proteins. PLoS Biol 4:1115–1121
Gao X, Liu X, Zhang Y, Wei Y, Wang Y (2020) Rapid and visual detection of porcine deltacoronavirus by recombinase polymerase amplification combined with a lateral flow dipstick. BMC Vet Res 16:130
Lau YL, Ismail IB, Mustapa NIB, Lai MY, Soh TST, Hassan AH, Peariasamy KM, Lee YL, Kahar MKBA, Chong JJ (2021) Development of a reverse transcription recombinase polymerase amplification assay for rapid and direct visual detection of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). PLoS ONE 16:e0245164
Miao F, Zhang J, Li N, Chen T, Wang L, Zhang F, Mi L, Zhang J, Wang S, Wang Y (2019) Rapid and sensitive recombinase polymerase amplification combined with lateral flow strip for detecting african swine fever virus. Front Microbiol 10:1004
Wang HM, Zhao GM, Hou PL, Yu L, He CQ, He HB (2018) Rapid detection of foot-and-mouth disease virus using reverse transcription recombinase polymerase amplification combined with a lateral flow dipstick. J Virol Methods 261:46–50
Yang Y, Qin X, Sun Y, Cong G, Li Y, Zhang Z (2017) Development of isothermal recombinase polymerase amplification assay for rapid detection of porcine circovirus type 2. Biomed Res Int 2017:1–8
Yu F, Yan Y, Shi M, Liu HZ, Zhang HL, Yang YB, Huang XY, Gauger PC, Zhang J, Zhang YH, Tong GZ, Tian ZJ, Chen JJ, Cai XH, Liu D, Li G, An TQ (2020) Phylogenetics, genomic recombination, and nsp2 polymorphic patterns of porcine reproductive and respiratory syndrome virus in China and the united states in 2014-2018. J Virol 94:e01813-e1819
Zhao K, Gao JC, Xiong JY, Guo JC, Yang YB, Jiang CG, Tang YD, Tian ZJ, Cai XH, Tong GZ, An TQ (2018) Two residues in NSP9 contribute to the enhanced replication and pathogenicity of highly pathogenic porcine reproductive and respiratory syndrome virus. J Virol 92:e02209-e2217
Chen N, Ye M, Xiao Y, Li S, Zhu JT (2019) Development of universal and quadruplex real-time RT-PCR assays for simultaneous detection and differentiation of porcine reproductive and respiratory syndrome viruses. Transbound Emerg Dis 66(6):2271–2278
Croft E, Blackwell T, Zimmerman J (2020) Field application of a commercial porcine reproductive and respiratory syndrome virus (PRRSV) oral fluid antibody enzyme-linked immunosorbent assay (ELISA). Can Vet J La revue veterinaire canadienne 61(4):420–423
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K (2000) Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28(12):E63
Compton JJ (1991) Nucleic acid sequence-based amplification. Mol Methods Virus Detect 28(28):261–285
Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC, Ward DC (1998) Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat Genet 19:225–232
Park JY, Park S, Park YR, Kang DY, Kim EM, Jeon HS, Kim JJ, Kim WI, Lee KT, Kim SH, Lee KK (2016) Reverse-transcription loop-mediated isothermal amplification (RT-LAMP) assay for the visual detection of European and North American porcine reproductive and respiratory syndrome viruses. J Virol Methods 237:10–13
Daher RK, Stewart G, Boissinot MG (2016) Recombinase polymerase amplification for diagnostic applications. Clin Chem 62:947–958
Yang Y, Qin X, Sun Y, Chen T, Zhang ZJVG (2016) Rapid detection of highly pathogenic porcine reproductive and respiratory syndrome virus by a fluorescent probe-based isothermal recombinase polymerase amplification assay. Virus Genes 52:883–886
Acknowledgements
The study was supported by grants from the National Natural Science Foundation of China (32072851, 31941019) and the State Key Laboratory of Veterinary Biotechnology Research Fund (SKLVBF202110, SKLVBF201902).
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: William G Dundon.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tian, XX., Wang, T., Cui, XY. et al. Rapid visual detection of porcine reproductive and respiratory syndrome virus via recombinase polymerase amplification combined with a lateral flow dipstick. Arch Virol 167, 493–499 (2022). https://doi.org/10.1007/s00705-021-05349-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-05349-8