
Conducting the RBD of SARS-CoV-2 Omicron Variant with Phytoconstituents from Euphorbia dendroides to Repudiate the Binding of Spike Glycoprotein Using Computational Molecular Search and Simulation Approach

 , ,
, ,  ,
,  , ,
, ,  , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Identification of Phytoconstituents from E. dendroides
2.2. Molecular Docking
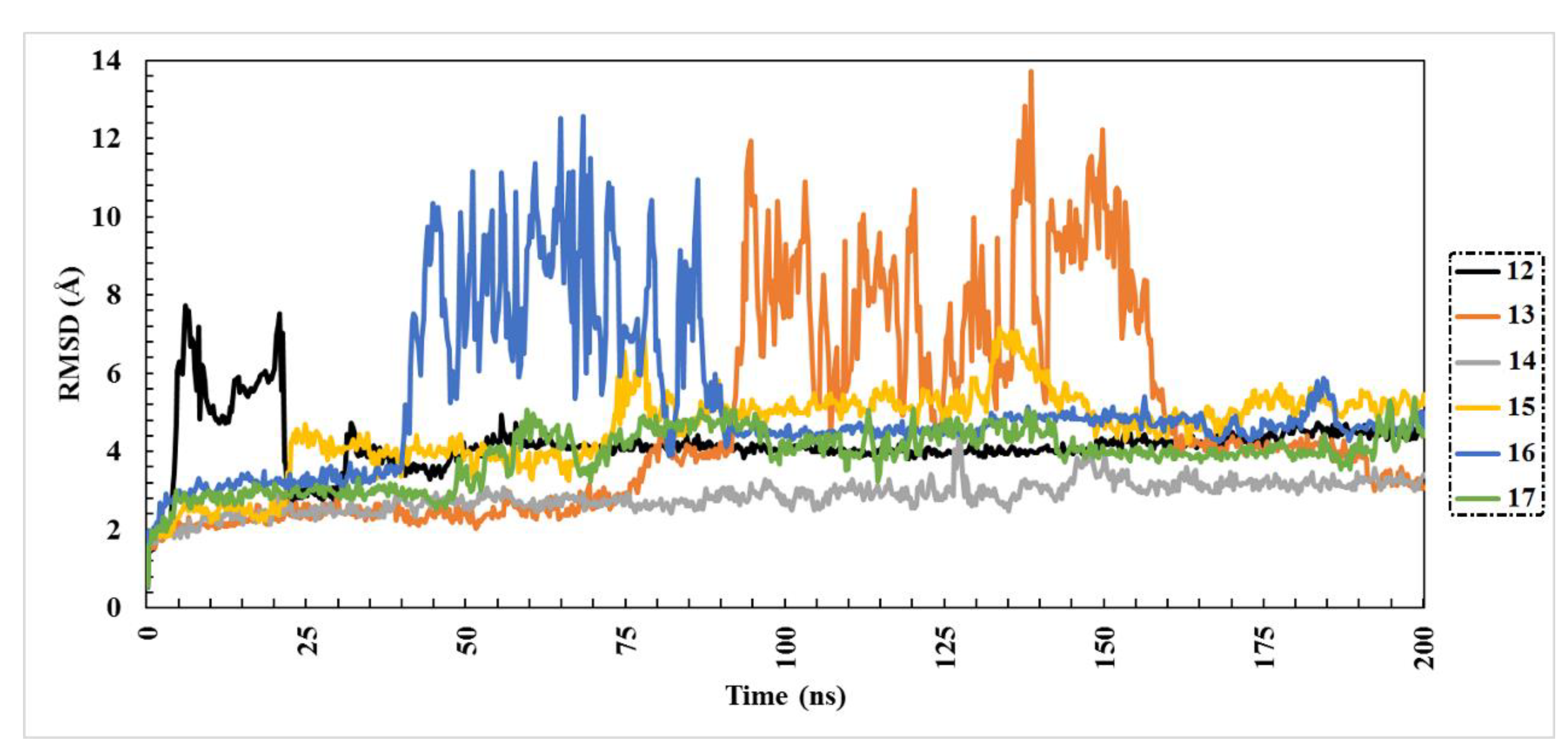
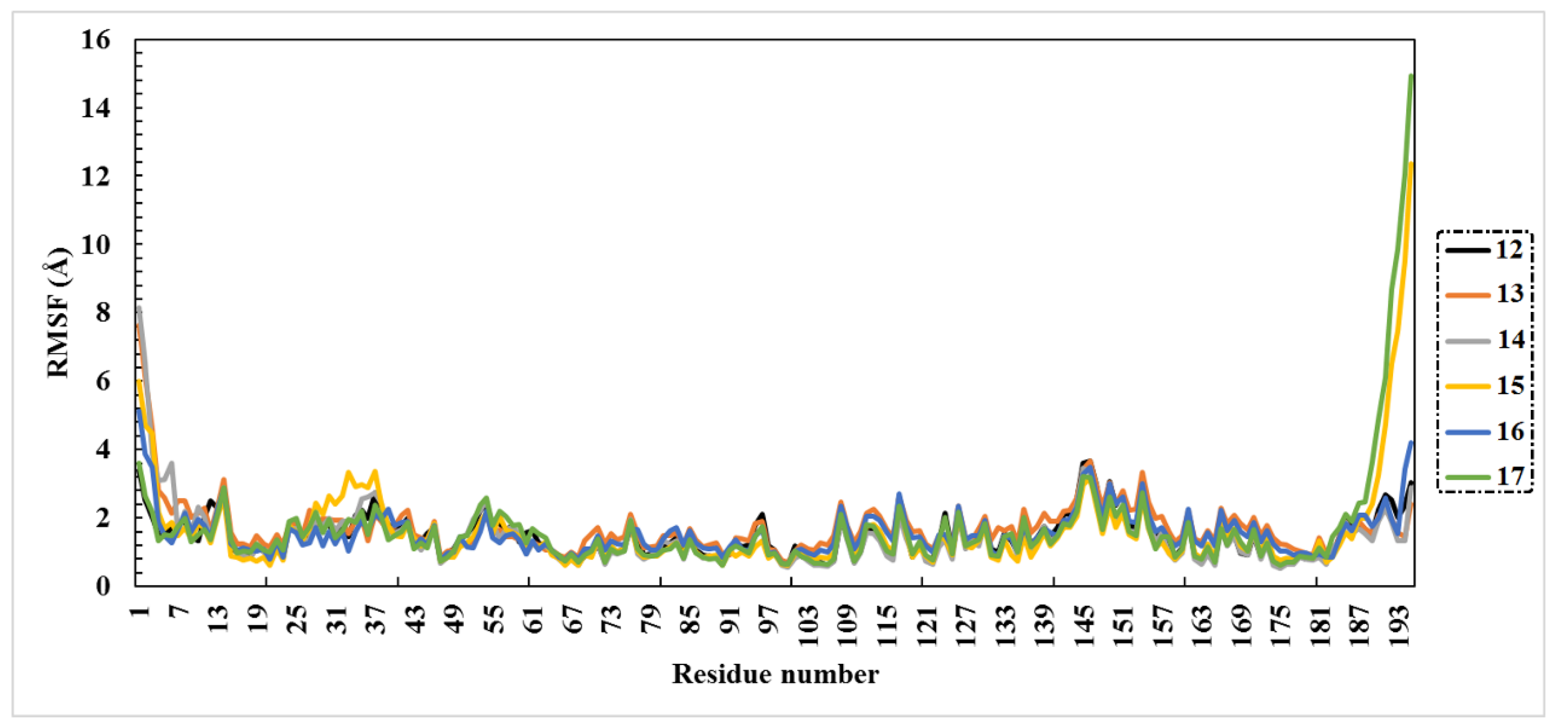
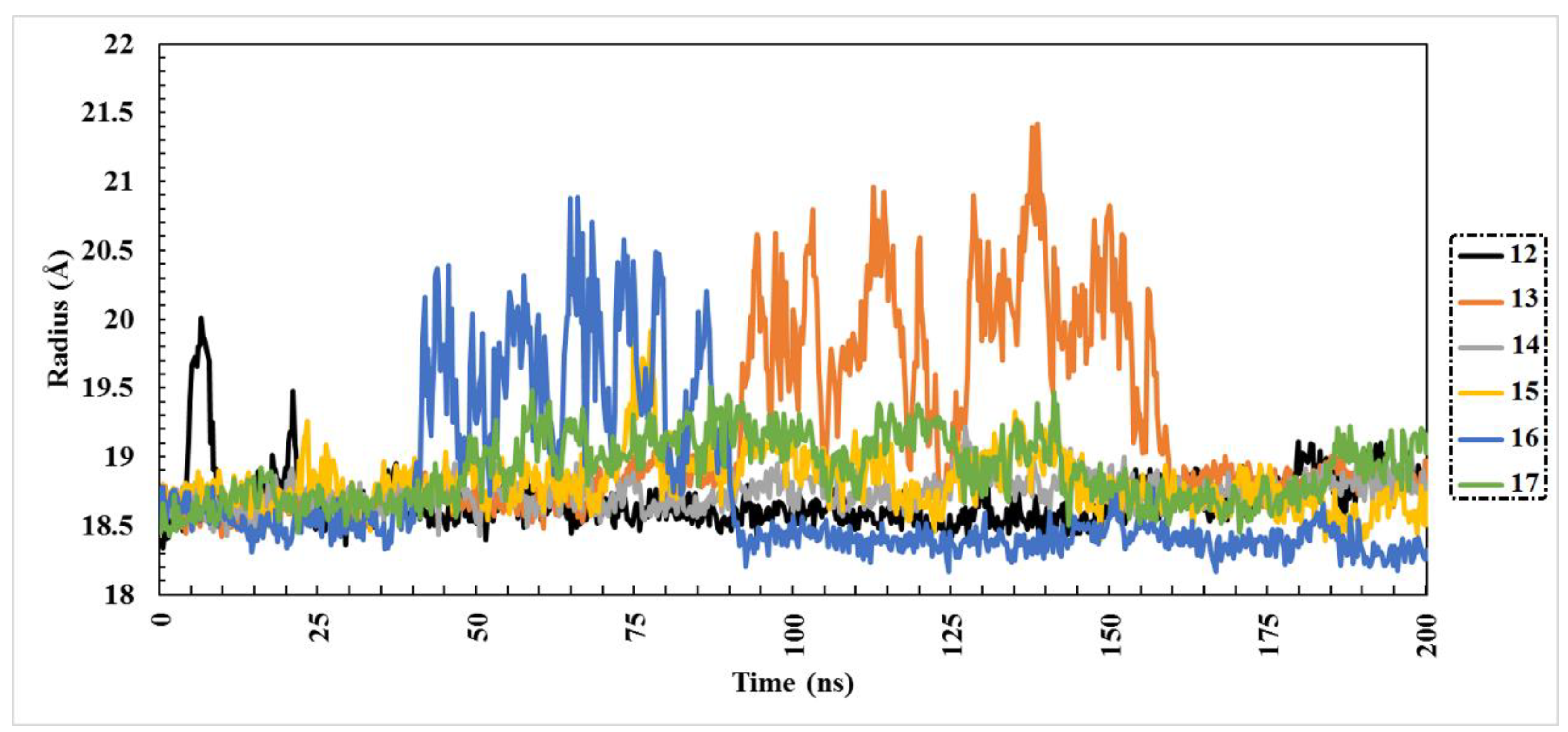
2.3. Molecular Dynamics (MD) Simulations
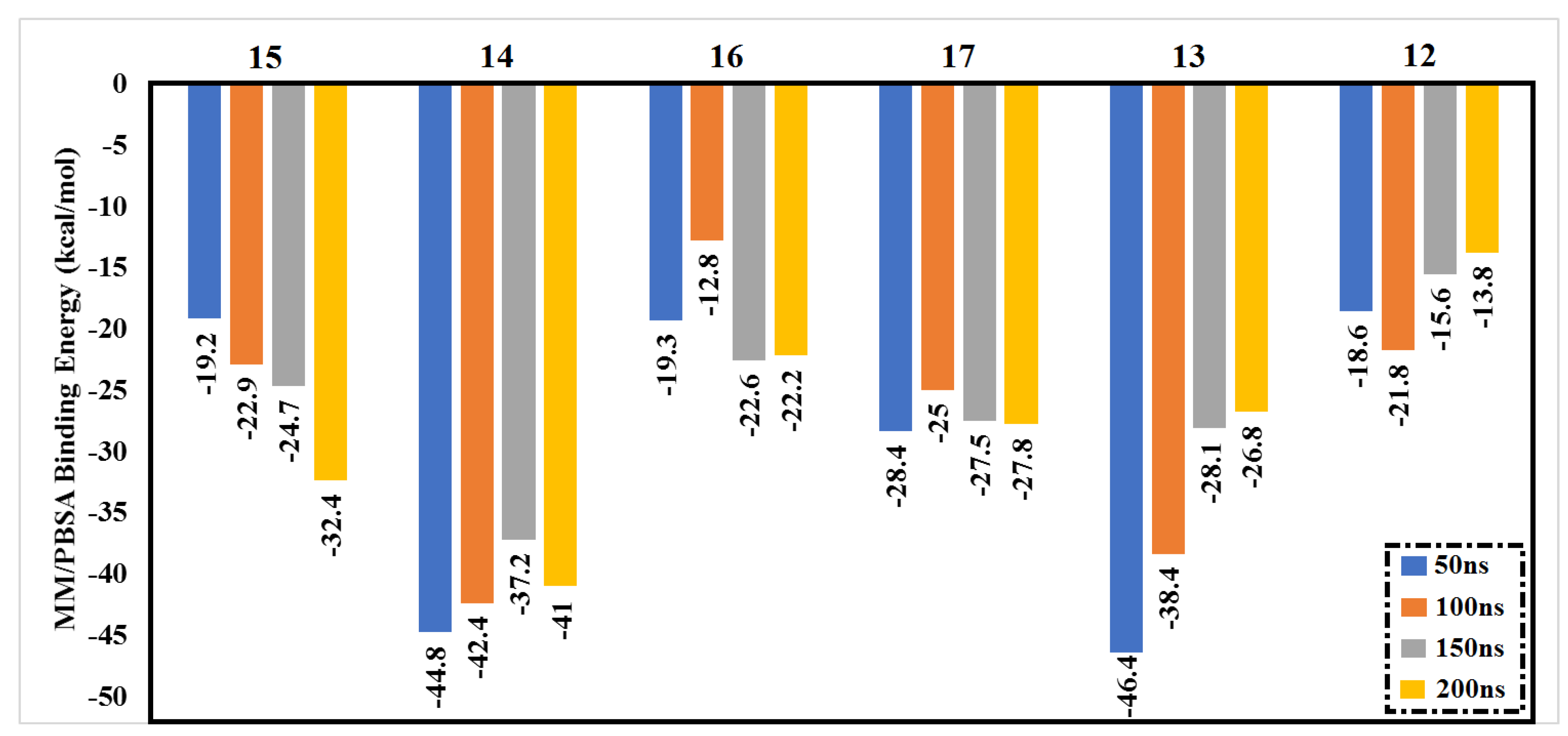
2.4. Post-MD Analyses
2.5. In Silico Drug Likeness
3. Materials and Methods
3.1. Plant Material
3.2. Phytochemical Constituents of E. dendroides
3.3. Protein Preparation
3.4. Inhibitor Preparation
3.5. Molecular Docking
3.6. Molecular Dynamics Simulations
3.7. Binding Energy Calculations
3.8. Drug Likeness Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Horne, J.R.; Vohl, M.-C. Biological plausibility for interactions between dietary fat, resveratrol, ACE2, and SARS-CoV illness severity. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E830–E833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, M.A.; Mohamed, E.A.; Abdelrahman, A.H.; Allemailem, K.S.; Moustafa, M.F.; Shawky, A.M.; Mahzari, A.; Hakami, A.R.; Abdeljawaad, K.A.; Atia, M.A. Rutin and flavone analogs as prospective SARS-CoV-2 main protease inhibitors: In silico drug discovery study. J. Mol. Graph. Model. 2021, 105, 107904. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, W.K.; Ha, A.W. Effects of Phytochemicals on Blood Pressure and Neuroprotection Mediated Via Brain Renin-Angiotensin System. Nutrients 2019, 11, 2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.-Y.; Yang, M.; Li, Z.; Meng, Z. Curcumin inhibits angiotensin II-induced inflammation and proliferation of rat vascular smooth muscle cells by elevating PPAR-γ activity and reducing oxidative stress. Int. J. Mol. Med. 2017, 39, 1307–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Sk, M.F.; Sonawane, A.; Kar, P.; Sadhukhan, S. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA-dependent RNA polymerase (RdRp) inhibition: An in-silico analysis. J. Biomol. Struct. Dyn. 2021, 39, 6249–6264. [Google Scholar] [CrossRef] [PubMed]
- Hepper, F.N. Flora of Egypt; Hardback, Al Hadara: Cairo, Egypt, 2001; Volume 1, ISBN 9789775429148. [Google Scholar]
- Ernst, M.; Grace, O.M.; Saslis-Lagoudakis, H.; Nilsson, N.; Simonsen, H.T.; Rønsted, N. Global medicinal uses of Euphorbia L. (Euphorbiaceae). J. Ethnopharmacol. 2015, 176, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Kemboi, D.; Peter, X.; Langat, M.; Tembu, J. A review of the ethnomedicinal uses, biological activities, and triterpenoids of Euphorbia species. Molecules 2020, 25, 4019. [Google Scholar] [CrossRef]
- Kumar, S.; Malhotra, R.; Kumar, D. Euphorbia hirta: Its chemistry, traditional and medicinal uses, and pharmacological activities. Pharmacogn. Rev. 2010, 4, 58–61. [Google Scholar] [CrossRef] [Green Version]
- Ghanadian, M.; Saeidi, H.; Aghaei, M.; Rahiminejad, M.R.; Ahmadi, E.; Ayatollahi, S.M.; Choudhary, M.I.; Bahmani, B. New jatrophane diterpenes from Euphorbia osyridea with proapoptotic effects on ovarian cancer cells. Phytochem. Lett. 2015, 12, 302–307. [Google Scholar] [CrossRef]
- Hegazy, M.-E.F.; Hamed, A.R.; Ibrahim, M.A.A.; Talat, Z.; Reda, E.H.; Abdel-Azim, N.S.; Hammouda, F.M.; Nakamura, S.; Matsuda, H.; Haggag, E.G.; et al. Euphosantianane A–D: Antiproliferative Premyrsinane Diterpenoids from the Endemic Egyptian Plant Euphorbia Sanctae-Catharinae. Molecules 2018, 23, 2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.R.; Ashour, A.; Amen, Y.; Nagata, M.; El-Toumy, S.A.; Shimizu, K. A new cycloartane triterpene and other phytoconstituents from the aerial parts of Euphorbia dendroides. Nat. Prod. Res. 2020, 36, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Ghout, A.; Zellagui, A.; Gherraf, N.; Demirtas, I.; Sahin, Y.A.; Boukhenaf, M.; Lahouel, M.; Nieto, G.; Akkal, S. Antiproliferative and Antioxidant Activities of Two Extracts of the Plant Species Euphorbia dendroides L. Medicines 2018, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Nothias-Scaglia, L.-F.; Dumontet, V.; Neyts, J.; Roussi, F.; Costa, J.; Leyssen, P.; Litaudon, M.; Paolini, J. LC-MS2-Based dereplication of Euphorbia extracts with anti-Chikungunya virus activity. Fitoterapia 2015, 105, 202–209. [Google Scholar] [CrossRef]
- Esposito, M.; Nothias, L.-F.; Nedev, H.; Gallard, J.-F.; Leyssen, P.; Retailleau, P.; Costa, J.; Roussi, F.; Iorga, B.I.; Paolini, J.; et al. Euphorbia dendroides Latex as a Source of Jatrophane Esters: Isolation, Structural Analysis, Conformational Study, and Anti-CHIKV Activity. J. Nat. Prod. 2016, 79, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Parmar, G.; Shah, A.; Shah, S.; Seth, A.K. Identification of Bioactive Phytoconstituents from the Plant Euphorbia hirta as Potential Inhibitor of SARS-CoV-2: An In-Silico Approach. Biointerface Res. Appl. Chem. 2022, 12, 1385–1396. [Google Scholar]
- Cayona, R.; Creencia, E. Phytochemicals of Euphorbia hirta L. and Their Inhibitory Potential Against SARS-CoV-2 Main Protease. Front. Mol. Biosci. 2021, 8, 801401. [Google Scholar] [CrossRef]
- Khursheed, A.; Jain, V. Euphorbia hirta as a gold mine of high-value phytochemicals: A comprehensive review of its pharmacological activities and possible role against SARS-CoV-2. Biomed. Res. Therapy. 2022, 9, 4930–4949. [Google Scholar] [CrossRef]
- Hassan, A.R. Chemical profile and cytotoxic activity of a polyphenolic-rich fraction from Euphorbia dendroides aerial parts. S. Afr. J. Bot. 2022, 147, 332–339. [Google Scholar] [CrossRef]
- Hassan, A.R.; Sanad, I.M.; Allam, A.E.; Abouelela, M.E.; Sayed, A.M.; Emam, S.S.; El-Kousy, S.M.; Shimizu, K. Chemical constituents from Limonium tubiflorum and their in silico evaluation as potential antiviral agents against SARS-CoV-2. Rsc Adv. 2021, 11, 32346–32357. [Google Scholar] [CrossRef]
- Khairan, K.; Idroes, R.; Tallei, T.E.; Nasim, M.J.; Jacob, C. Bioactive Compounds from Medicinal Plants and their Possible Effect as Therapeutic Agents against COVID-19: A Review. Curr. Nutr. Food Sci. 2021, 17, 621–633. [Google Scholar] [CrossRef]
- Khan, M.T.H.; Khan, S.B.; Ather, A. Tyrosinase inhibitory cycloartane type triterpenoids from the methanol extract of the whole plant of Amberboa ramosa Jafri and their structure–activity relationship. Bioorganic Med. Chem. 2006, 14, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Escobedo-Martínez, C.; Lozada, M.C.; Hernández-Ortega, S.; Villarreal, M.L.; Gnecco, D.; Enríquez, R.G.; Reynolds, W. 1H and 13C NMR characterization of new cycloartane triterpenes from Mangifera indica. Org. Magn. Reson. 2012, 50, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.T.M.; Magalhães, C.G.; Duarte, L.P.; Mussel, W.d.N.; Ruiz, A.L.T.G.; Shiozawa, L.; Carvalho, J.E.d.; Trindade, I.C.; Vieira, S.A. Lupeol and its esters: NMR, powder XRD data and in vitro evaluation of cancer cell growth. Braz. J. Pharm. Sci. 2017, 53, e00251. [Google Scholar] [CrossRef] [Green Version]
- Ghani, A.; Badr, W. New Acetyl Triterpenoidal and Biological Activities of Euphorbia Paralias and Euophorbia Geniculata (Euphorbiaceae) from Egypt. Egypt. J. Chem. 2020, 63, 3583–3595. [Google Scholar]
- Ghanadian, M.; Sadraei, H.; Yousuf, S.; Asghari, G.; Choudhary, M.I.; Jahed, M. New diterpene polyester and phenolic compounds from Pycnocycla spinosa Decne. Ex Boiss with relaxant effects on KCl-induced contraction in rat ileum. Phytochem. Lett. 2014, 7, 57–61. [Google Scholar] [CrossRef]
- Hassan, A.R.; El-Kousy, S.M.; El-Toumy, S.A.; Frydenvang, K.; Tung, T.T.; Olsen, J.; Nielsen, J.; Christensen, S.B. Metformin, an Anthropogenic Contaminant of Seidlitzia rosmarinus Collected in a Desert Region near the Gulf of Aqaba, Sinai Peninsula. J. Nat. Prod. 2017, 80, 2830–2834. [Google Scholar] [CrossRef]
- Wei, Y.; Gao, Y.; Zhang, K.; Ito, Y. Isolation of caffeic acid from eupatorium adenophorum spreng by high-speed countercurrent chromatography and syn-thesis of caffeic acid-intercalated layered double hydroxide. J. Liq. Chromatogr. Relat. Technol. 2010, 33, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Atta, E.M.; Nassar, A.A.; Hasan, N.M.; Hasan, A.R. New Flavonoid Glycoside and Pharmacological Activities of Pteranthus dichotomus Forssk. Rec. Nat. Prod. 2013, 7, 69–79. [Google Scholar]
- Ahmed, H.; Moawad, A.; Owis, A.; AbouZid, S.; Ahmed, O. Flavonoids of Calligonum polygonoides and their cytotoxicity. Pharm. Biol. 2016, 54, 2119–2126. [Google Scholar] [CrossRef] [Green Version]
- Rezende, F.M.; Ferreira, M.J.P.; Clausen, M.H.; Rossi, M.; Furlan, C.M. Acylated Flavonoid Glycosides are the Main Pigments that Determine the Flower Colour of the Brazilian Native Tree Tibouchina pulchra (Cham.) Cogn. Molecules 2019, 24, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magozwi, D.; Dinala, M.; Mokwana, N.; Siwe-Noundou, X.; Krause, R.; Sonopo, M.; McGaw, L.; Augustyn, W.; Tembu, V. Flavonoids from the Genus Euphorbia: Isolation, Structure, Pharmacological Activities and Structure–Activity Relationships. Pharmaceuticals 2021, 14, 428. [Google Scholar] [CrossRef] [PubMed]
- Vangeel, L.; Chiu, W.; De Jonghe, S.; Maes, P.; Slechten, B.; Raymenants, J.; André, E.; Leyssen, P.; Neyts, J.; Jochmans, D. Remdesivir, Molnupiravir and Nirmatrelvir remain active against SARS-CoV-2 Omicron and other variants of concern. Antivir. Res. 2022, 198, 105252. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Mullard, A. Re-assessing the rule of 5, two decades on. Nat. Rev. Drug Discov. 2018, 17, 777. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p K as and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [Green Version]
- Allinger, N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA, D.S. BIOVIA Discovery Studio Visualizer. Software Version. 2017. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 6 April 2022).
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 1986, 33, 3628–3631. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Reher, R.; Kühl, T.; Annala, S.; Benkel, T.; Kaufmann, D.; Nubbemeyer, B.; Odhiambo, J.P.; Heimer, P.; Bäuml, C.A.; Kehraus, S.; et al. Deciphering Specificity Determinants for FR900359-Derived Gqα Inhibitors Based on Computational and Structure-Activity Studies. ChemMedChem 2018, 13, 1634–1643. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-Limited Steps of Human Oral Absorption and QSAR Studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | 2D Chemical Structure | Docking Score (kcal/mol) | Binding Features (Hydrogen Bond Length in Å) |
|---|---|---|---|
| 15 |  | −8.8 | ARG403 (3.23 Å), TYR453 (2.94 Å), SER496 (2.99, 3.01 Å), TYR501 (2.92 Å), HIS505 (3.17 Å) |
| 14 |  | −8.7 | ARG403 (3.18 Å), SER496 (2.13, 2.93, 3.01 Å), TYR501 (2.89 Å), HIS505 (3.18 Å) |
| 16 |  | −8.4 | GLU406 (2.94 Å), TYR453 (2.87 Å), SER496 (2.92, 3.01 Å), TYR501 (2.90 Å), HIS505 (3.17 Å) |
| 17 |  | −8.3 | TYR453 (2.97 Å), SER496 (2.99, 3.03, 3.08 Å), TYR501 (2.94 Å), HIS505 (3.17 Å) |
| 13 |  | −8.1 | TYR453 (2.26 Å), SER496 (2.93, 2.98, 3.01 Å), TYR501 (2.87 Å), HIS505 (3.15 Å) |
| Remdesivir |  | −8.0 | TYR453 (2.91 Å), SER494 (2.27, 2.89 Å), TYR501 (3.01 Å) |
| 12 |  | −7.9 | ARG403 (3.04 Å), TYR495 (2.57 Å) |
| Molecule | miLogP | TPSA | MWt | nON | nOHNH | Nrotb | Nviolations | %ABS |
|---|---|---|---|---|---|---|---|---|
| 14 | −0.5 | 227.6 | 478.4 | 13 | 8 | 4 | 2 | 30.5% |
| 15 | 0.1 | 216.6 | 492.4 | 13 | 7 | 5 | 2 | 34.3% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, H.A.; Hassan, A.R.; Mohamed, E.A.R.; Al-Khdhairawi, A.; Karkashan, A.; Attar, R.; Allemailem, K.S.; Al Abdulmonem, W.; Shimizu, K.; Abdel-Rahman, I.A.M.; et al. Conducting the RBD of SARS-CoV-2 Omicron Variant with Phytoconstituents from Euphorbia dendroides to Repudiate the Binding of Spike Glycoprotein Using Computational Molecular Search and Simulation Approach. Molecules 2022, 27, 2929. https://doi.org/10.3390/molecules27092929
Hassan HA, Hassan AR, Mohamed EAR, Al-Khdhairawi A, Karkashan A, Attar R, Allemailem KS, Al Abdulmonem W, Shimizu K, Abdel-Rahman IAM, et al. Conducting the RBD of SARS-CoV-2 Omicron Variant with Phytoconstituents from Euphorbia dendroides to Repudiate the Binding of Spike Glycoprotein Using Computational Molecular Search and Simulation Approach. Molecules. 2022; 27(9):2929. https://doi.org/10.3390/molecules27092929
Chicago/Turabian StyleHassan, Heba Ali, Ahmed R. Hassan, Eslam A.R. Mohamed, Ahmad Al-Khdhairawi, Alaa Karkashan, Roba Attar, Khaled S. Allemailem, Waleed Al Abdulmonem, Kuniyoshi Shimizu, Iman A. M. Abdel-Rahman, and et al. 2022. "Conducting the RBD of SARS-CoV-2 Omicron Variant with Phytoconstituents from Euphorbia dendroides to Repudiate the Binding of Spike Glycoprotein Using Computational Molecular Search and Simulation Approach" Molecules 27, no. 9: 2929. https://doi.org/10.3390/molecules27092929