
Differential Effect of SARS-CoV-2 Spike Glycoprotein 1 on Human Bronchial and Alveolar Lung Mucosa Models: Implications for Pathogenicity

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Recombinant Spike Glycoprotein S1
2.2. Bronchial and Alveolar Lung Mucosal Models:
2.2.1. Bronchial
2.2.2. Alveolar
2.3. Exposure to Spike Protein
2.4. Cytotoxicity Assessment
2.5. Surface Expression of ACE2, TLR2, and TLR4
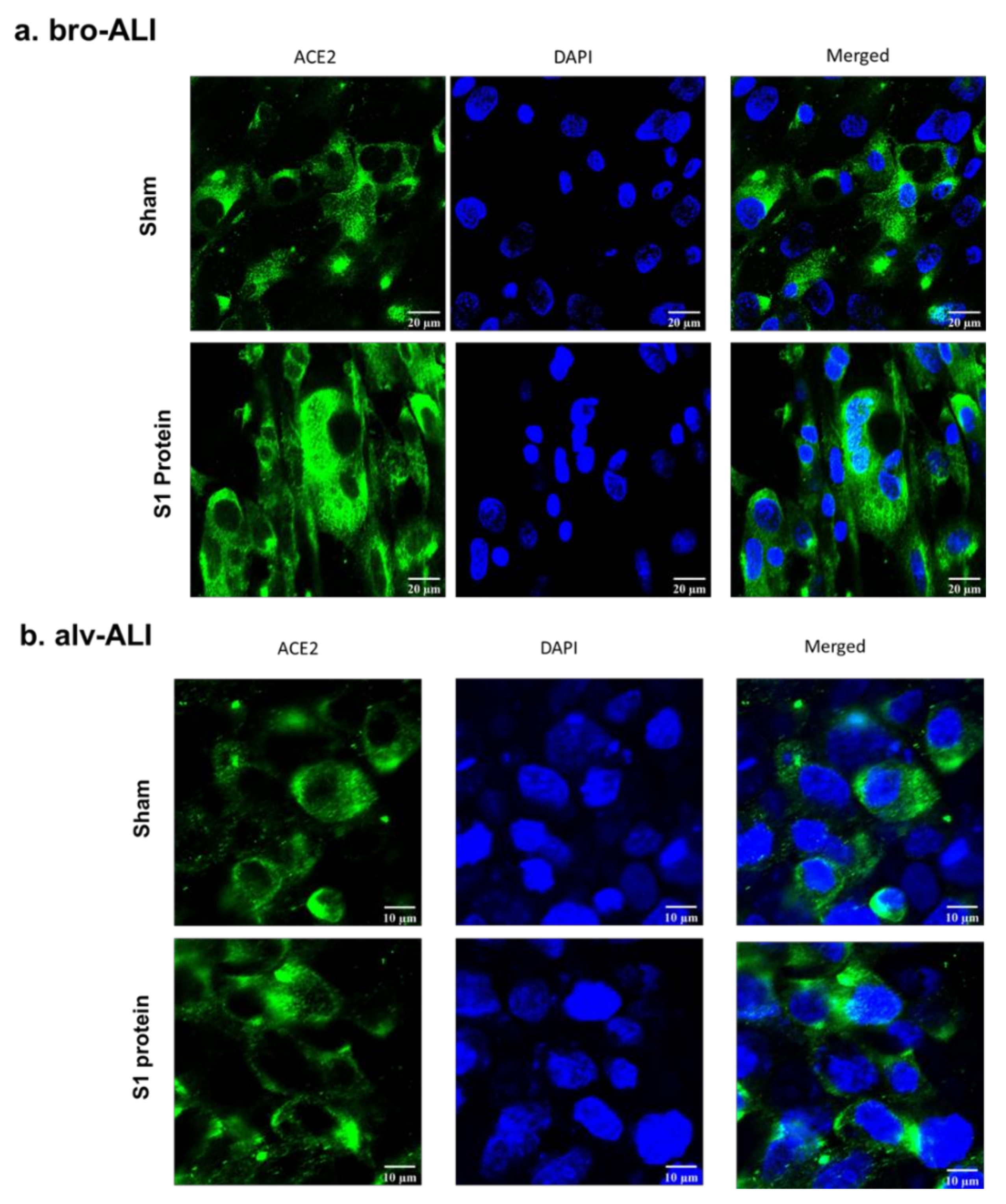
2.6. Confocal Microscopy of ACE2 Expression
2.7. Transcriptomic Analysis
2.8. Secreted Cytokine Concentration
2.9. Statistics
3. Results
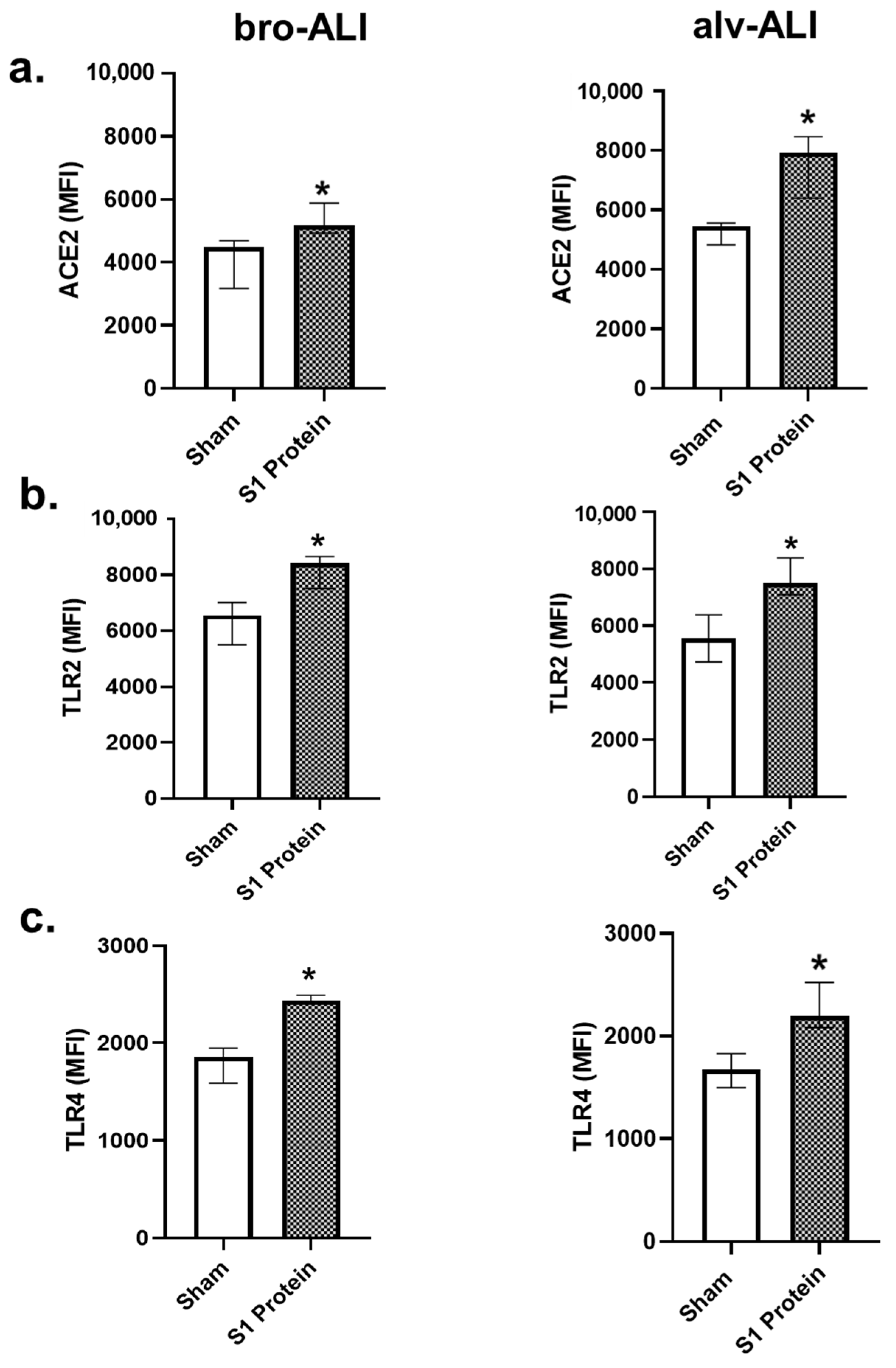
3.1. Increased ACE2, TLR2, and TLR4 Surface Expression
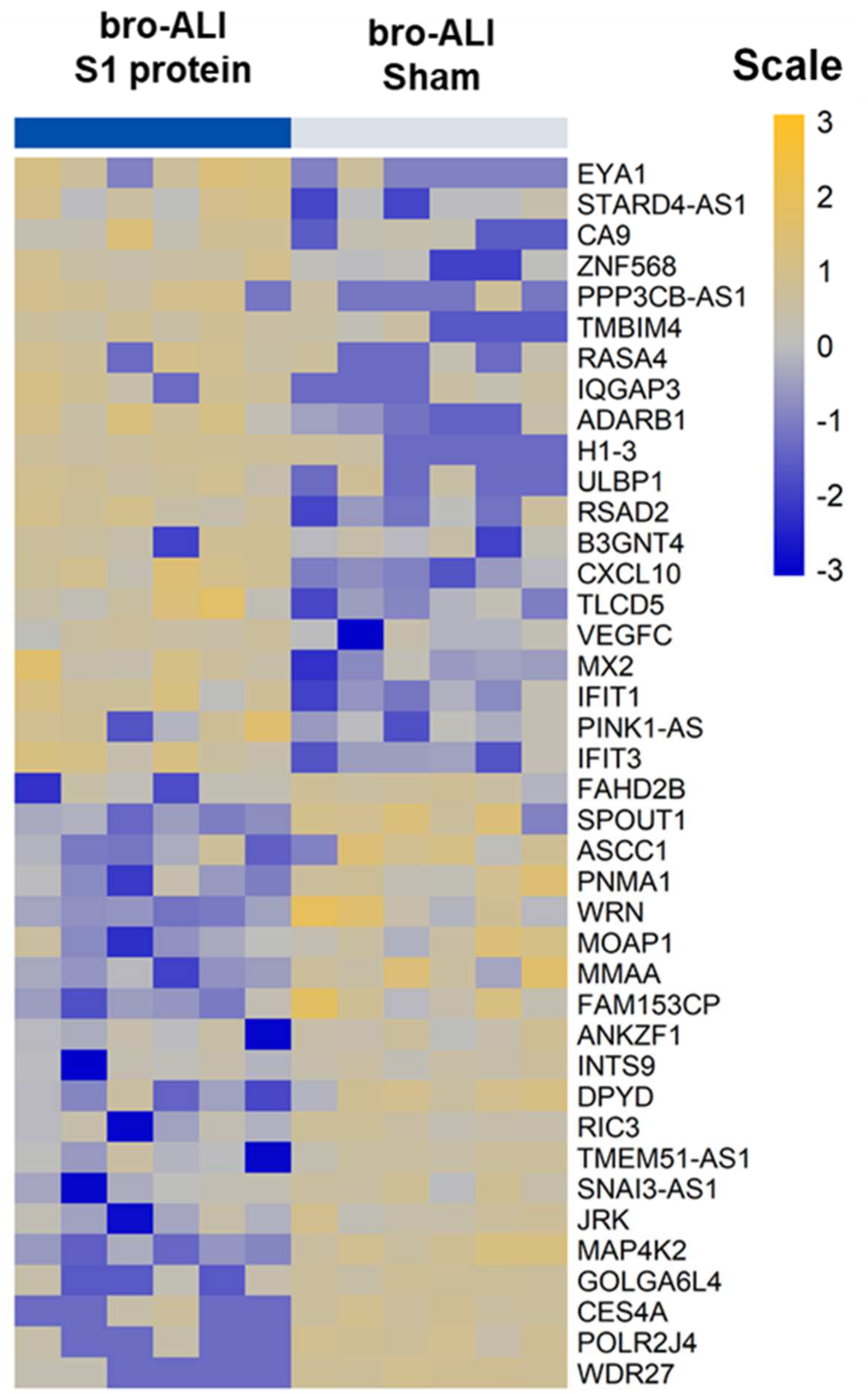
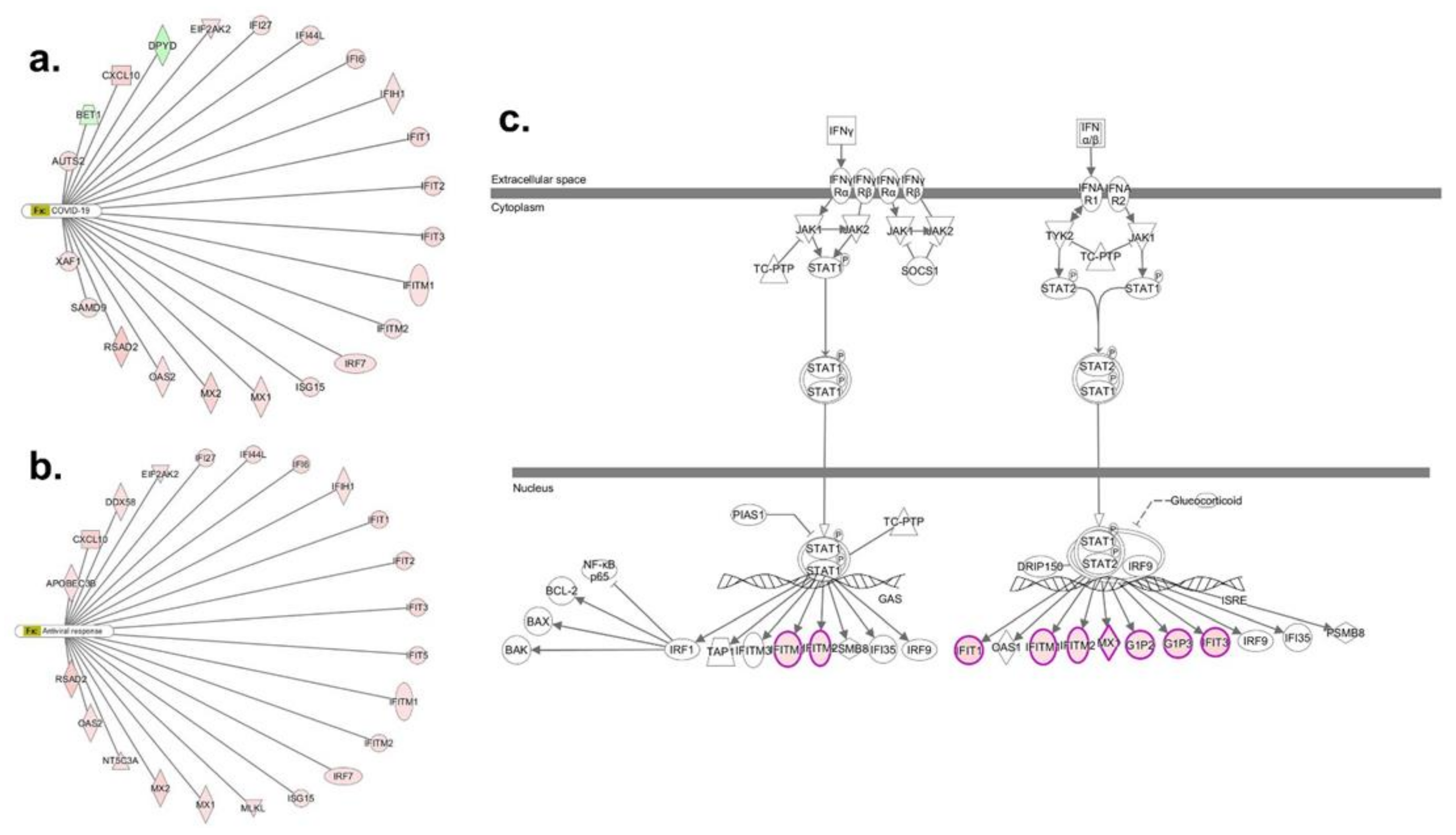
3.2. Transcriptomic Response in bro-ALI

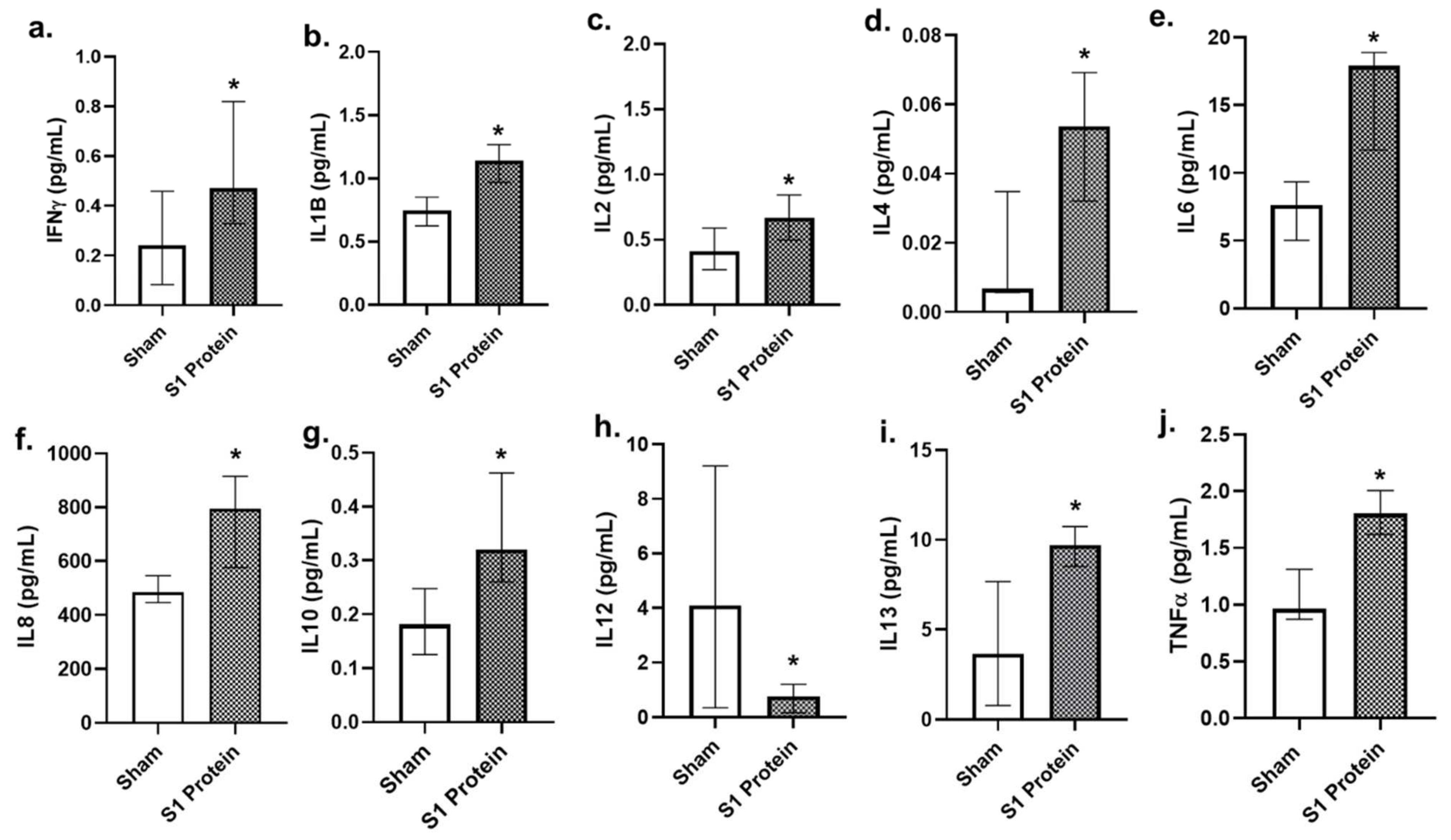
3.3. Secreted Cytokines in bro-ALI
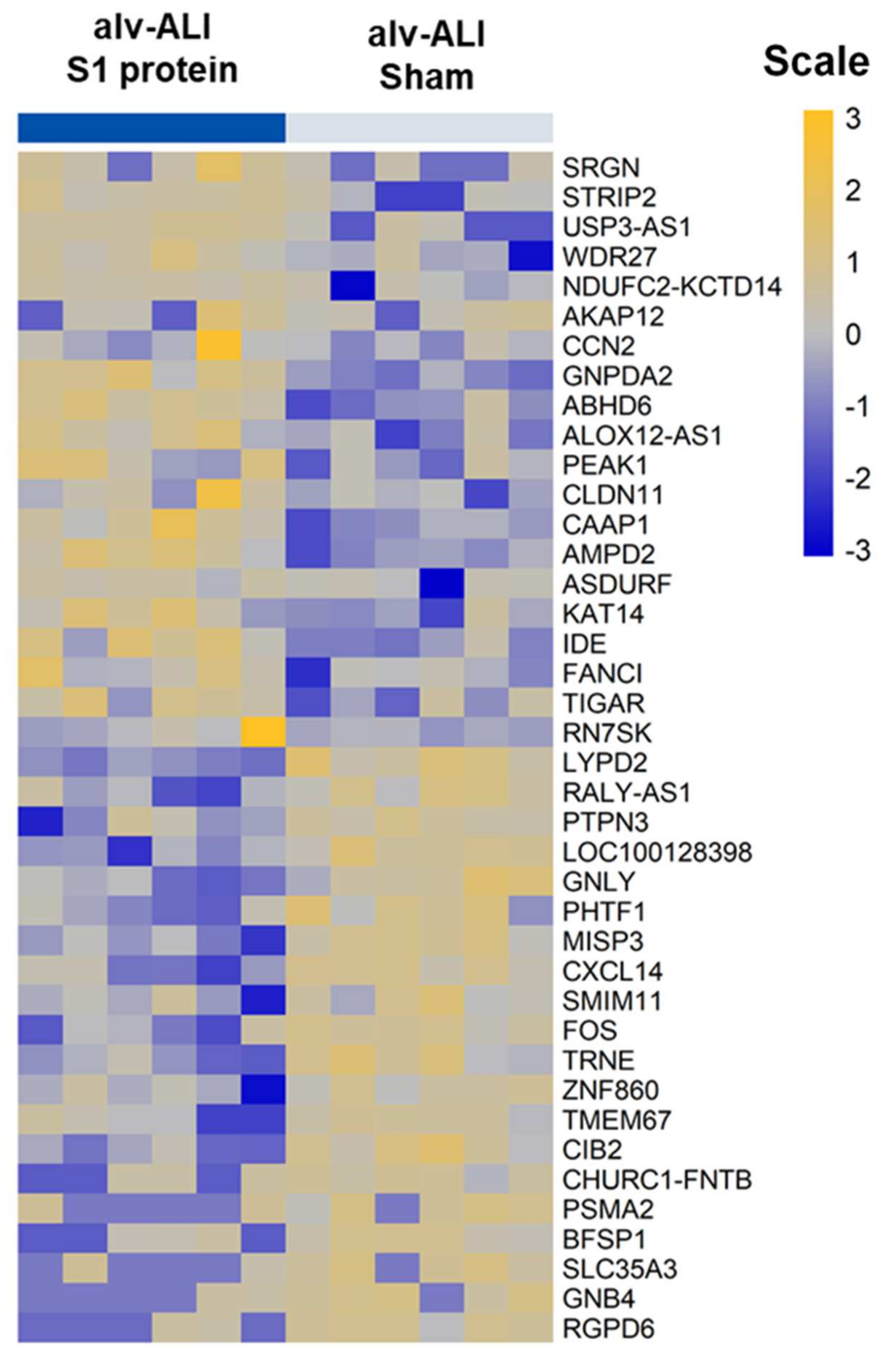
3.4. Transcriptomic Response in alv-ALI

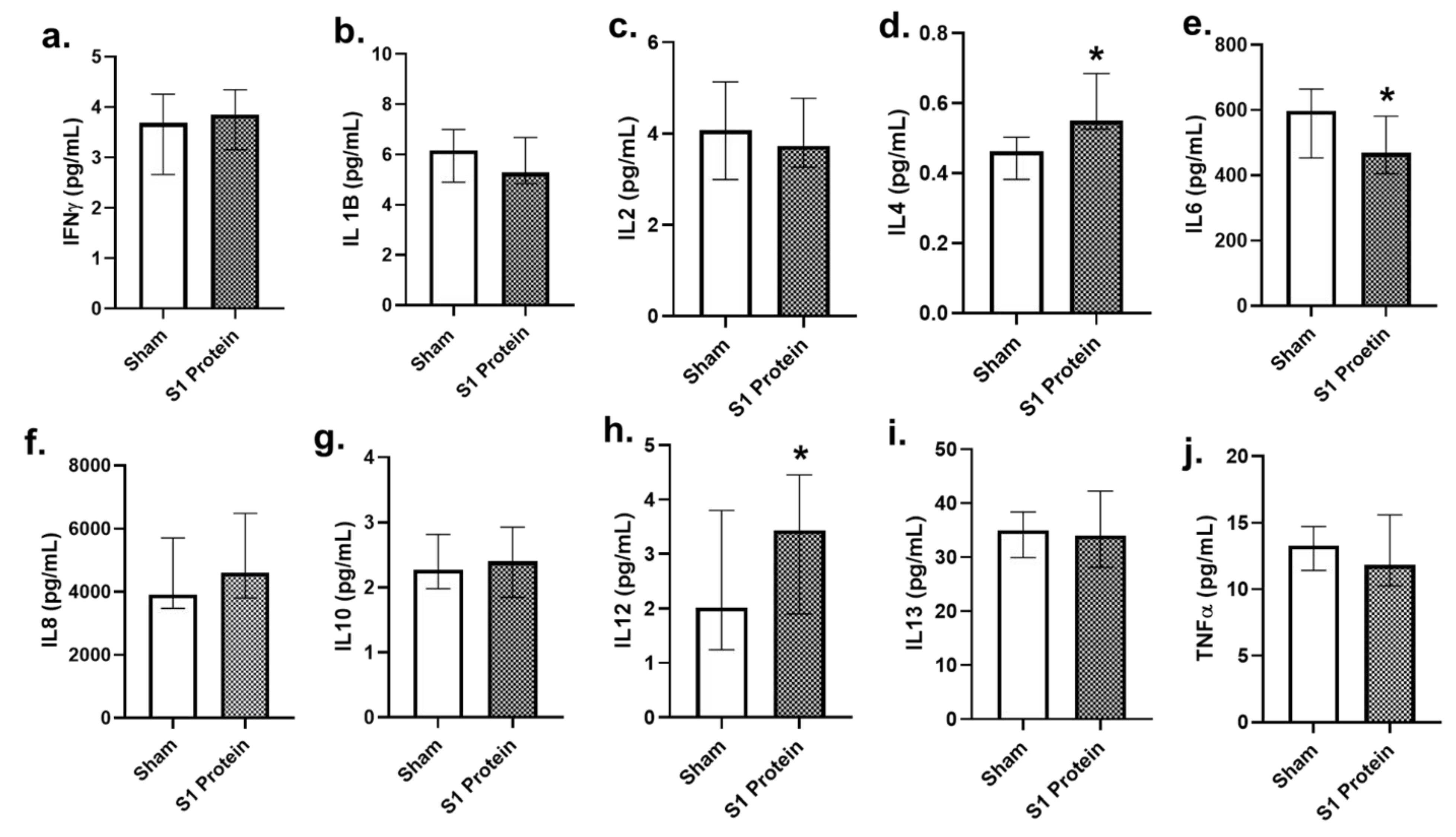
3.5. Secreted Cytokines in alv-ALI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harvey, W.T.C.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Suzuki, Y.J.G.; Gychka, S.G. SARS-CoV-2 Spike Protein Elicits Cell Signaling in Human Host Cells: Implications for Possible Consequences of COVID-19 Vaccines. Vaccines 2021, 9, 36. [Google Scholar] [CrossRef]
- ECDC. 2021. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 16 November 2021).
- WHO. Tracking SARS-CoV-2. Variants. 2021. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 16 November 2021).
- Lee, I.T.; Nakayama, T.; Wu, C.T.; Goltsev, Y.; Jiang, S.; Gall, P.A.; Liao, C.K.; Shih, L.C.; Schürch, C.M.; McIlwain, D.R.; et al. ACE2 localizes to the respiratory cilia and is not increased by ACE inhibitors or ARBs. Nat. Commun. 2020, 11, 5453. [Google Scholar] [CrossRef]
- Wijnant, S.R.; Jacobs, M.; Eeckhoutte, H.P.V.; Lapauw, B.; Joos, G.F.; Bracke, K.R.; Brusselle, G.G. Expression of ACE2, the SARS-CoV-2 Receptor, in Lung Tissue of Patients With Type 2 Diabetes. Diabetes 2020, 69, 2691–2699. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, N.Z.; Grandvaux, N. ACE2: Evidence of role as entry receptor for SARS-CoV-2 and implications in comorbidities. Elife 2020, 9, e61390. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Geri, P.; Salton, F.; Zuccatosta, L.; Tamburrini, M.; Tamburrini, M.; Biolo, M.; Busca, A.; Santagiuliana, M.; Zuccon, U.; Confalonieri, P.; et al. Limited role for bronchoalveolar lavage to exclude COVID-19 after negative upper respiratory tract swabs: A multicentre study. Eur. Respir. J. 2020, 56, 2001733. [Google Scholar] [CrossRef]
- Voiriot, G.; Fajac, A.; Lopinto, J.; Labbé, V.; Fartoukh, M. Bronchoalveolar lavage findings in severe COVID-19 pneumonia. Intern. Emerg. Med. 2020, 15, 1333–1334. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Williams, E.P. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Sariol, A.; Perlman, S. SARS-CoV-2 takes its Toll. Nat. Immunol. 2021, 22, 801–802. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Rezaei, N. Role of Toll-like receptors in the pathogenesis of COVID-19. J. Med. Virol. 2021, 93, 2735–2739. [Google Scholar] [CrossRef] [PubMed]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Hemmat, N.; Asadzadeh, Z.; Ahangar, N.K.; Alemohammad, H.; Najafzadeh, B.; Derakhshani, A.; Baghbanzadeh, A.; Baghi, H.B.; Javadrashid, D.; Najafi, S.; et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021, 166, 675–696. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J.; Nikolaienko, S.I.; Dibrova, V.A.; Vasylyk, V.M.; Novikov, M.Y.; Shults, N.V.; Gychka, S.G. SARS-CoV-2 spike protein-mediated cell signaling in lung vascular cells. Vascul. Pharmacol. 2021, 137, 106823. [Google Scholar] [CrossRef]
- John, E.A.; Joseph, C.; Jenkins, G.; Tatler, A.L. COVID-19 and pulmonary fibrosis: A potential role for lung epithelial cells and fibroblasts. Immunol. Rev. 2021, 302, 228–240. [Google Scholar] [CrossRef]
- Ouyang, L.; Gong, J.; Yu, M. Pre-existing interstitial lung disease in patients with coronavirus disease 2019: A meta-analysis. Int. Immunopharmacol. 2021, 100, 108145. [Google Scholar] [CrossRef] [PubMed]
- Baratella, E.; Ruaro, B.; Marrocchio, C.; Starvaggi, N.; Salton, F.; Giudici, F.; Quaia, E.; Confalonieri, M.; Cova, M.A. Interstitial Lung Disease at High Resolution CT after SARS-CoV-2-Related Acute Respiratory Distress Syndrome According to Pulmonary Segmental Anatomy. J. Clin. Med. 2021, 10, 3985. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Hedelin, A.; Malmlöf, M.; Kessler, V.; Seisenbaeva, G.; Gerde, P.; Palmberg, L. Development of Combining of Human Bronchial Mucosa Models with XposeALI® for Exposure of Air Pollution Nanoparticles. PLoS ONE 2017, 12, e0170428. [Google Scholar] [CrossRef]
- Ji, J.; Ganguly, K.; Mihai, X.; Sun, J.; Malmlöf, M.; Gerde, P.; Upadhyay, S.; Palmberg, L. Exposure of normal and chronic bronchitis-like mucosa models to aerosolized carbon nanoparticles: Comparison of pro-inflammatory oxidative stress and tissue injury/repair responses. Nanotoxicology 2018, 13, 1362–1379. [Google Scholar] [CrossRef]
- Thimraj, T.A.; Sompa, S.I.; Ganguly, K.; Ernstgård, L.; Johanson, G.; Palmberg, L.; Upadhyay, S. Evaluation of diacetyl mediated pulmonary effects in physiologically relevant air-liquid interface models of human primary bronchial epithelial cells. Toxicol. In Vitro 2019, 61, 104617. [Google Scholar] [CrossRef]
- Ganguly, K.; Nordström, A.; Thimraj, T.A.; Rahman, M.; Ramström, M.; Sompa, S.I.; Lin, E.Z.; O’Brien, F.; Koelmel, J.; Ernstgård, L.; et al. Addressing the challenges of E-cigarette safety profiling by assessment of pulmonary toxicological response in bronchial and alveolar mucosa models. Sci. Rep. 2020, 10, 20460. [Google Scholar] [CrossRef] [PubMed]
- Dosch, S.F.; Mahajan, S.D.; Collins, A.R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 2009, 142, 19–27. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, w191–w198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. bioRxiv 2021. [CrossRef]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.B.; Prabhu, A.; Bhandary, Y.P. Targeting anti-aging protein sirtuin (Sirt) in the diagnosis of idiopathic pulmonary fibrosis. J. Cell. Biochem. 2018, 120, 6878–6885. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Fujimoto, M.; Hasegawa, M.; Tanaka, C.; Kumada, S.; Ogawa, F.; Takehara, K.; Sato, S. Elevated serum APRIL levels in patients with systemic sclerosis: Distinct profiles of systemic sclerosis categorized by APRIL and BAFF. J. Rheumatol. 2007, 10, 2056–2062. [Google Scholar]
- Kavvadas, P.; Kypreou, K.P.; Protopapadakis, E.; Prodromidi, E.; Sideras, P.; Charonis, A.S. Integrin-linked kinase (ILK) in pulmonary fibrosis. Virchows Arch. 2010, 457, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, L.A. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin. Immunopathol. 2016, 38, 517–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Li, Y.; Yu, J.; Dong, L.; Husain, A.N.; Shen, L.; Weber, C.R. Idiopathic pulmonary fibrosis is associated with tight junction protein alterations. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183205. [Google Scholar] [CrossRef]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Toews, G.B. Cytokines and the lung. Eur. Respir. J. Suppl. 2001, 34, 3s–17s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, S.; Palmberg, L. Air-Liquid Interface: Relevant In Vitro Models for Investigating Air Pollutant-Induced Pulmonary Toxicity. Toxicol. Sci. 2018, 164, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, Y.X.; Yu, Q.Y.; Tang, X.X. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discov. 2021, 7, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Gasse, P.; Mary, C.; Guenon, I.; Noulin, N.; Charron, S.; Schnyder-Candrian, S.; Schnyder, B.; Akira, S.; Quesniaux, V.F.J.; Lagente, V.; et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. Investig. 2007, 117, 3786–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lee, T.C.; Guillemin, B.; Yu, M.C.; Rom, W.N. Enhanced IL-1 beta and tumor necrosis factor-alpha release and messenger RNA expression in macrophages from idiopathic pulmonary fibrosis or after asbestos exposure. J. Immunol. 1993, 150, 4188–4196. [Google Scholar]
- Kline, J.N.; Schwartz, D.A.; Monick, M.M.; Floerchinger, C.S.; Hunninghake, G.W. Relative release of interleukin-1 beta and interleukin-1 receptor antagonist by alveolar macrophages. A study in asbestos-induced lung disease, sarcoidosis, and idiopathic pulmonary fibrosis. Chest 1993, 104, 47–53. [Google Scholar] [CrossRef]
- Gillery, P.; Fertin, C.; Nicolas, J.F.; Chastang, F.; Kalis, B.; Banchereau, J.; Maquart, F.X. Interleukin-4 stimulates collagen gene expression in human fibroblast monolayer cultures. Potential role in fibrosis. FEBS Lett. 1992, 302, 231–234. [Google Scholar] [CrossRef] [Green Version]
- Sempowski, G.D.; Derdak, S.; Phipps, R.P. Interleukin-4 and interferon-gamma discordantly regulate collagen biosynthesis by functionally distinct lung fibroblast subsets. J. Cell. Physiol. 1996, 167, 290–296. [Google Scholar] [CrossRef]
- Moodley, Y.P.; Misso, N.L.; Scaffidi, A.K.; Fogel-Petrovic, M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am. J. Respir. Cell Mol. Biol. 2003, 29, 490–498. [Google Scholar] [CrossRef]
- Moodley, Y.P.; Scaffidi, A.K.; Misso, N.L.; Keerthisingam, C.; McAnulty, R.J.; Laurent, G.J.; Mutsaers, S.E.; Thompson, P.J.; Knight, D.A. Fibroblasts isolated from normal lungs and those with idiopathic pulmonary fibrosis differ in interleukin-6/gp130-mediated cell signaling and proliferation. Am. J. Pathol. 2003, 163, 345–354. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, R.J.; Knight, D.A.; Richards, C.D.; Prêle, C.M.; Lau, H.L.; Jarnicki, A.G.; Jones, J.; Bozinovski, S.; Vlahos, R.; Thiem, S. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 2012, 4, 939–951. [Google Scholar] [CrossRef]
- Yang, L.; Herrera, J.; Gilbertsen, A.; Xia, H.; Smith, K.; Benyumov, A.; Bitterman, P.B.; Henke, C.A. IL-8 mediates idiopathic pulmonary fibrosis mesenchymal progenitor cell fibrogenicity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L127–L136. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, F.; Uchibori, R.; Sehara, Y.; Saga, Y.; Urabe, M.; Mizukami, H.; Hagiwara, K.; Kume, A. AAV6-Mediated IL-10 Expression in the Lung Ameliorates Bleomycin-Induced Pulmonary Fibrosis in Mice. Hum. Gene Ther. 2018, 29, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Louie, M.C.; Vannella, K.M.; Wilke, C.A.; LeVine, A.M.; Moore, B.B.; Shanley, T.P. New concepts of IL-10-induced lung fibrosis: Fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L341–L353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, M.P.; Belperio, J.A.; Burdick, M.D.; Strieter, R.M. IL-12 attenuates bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L92–L97. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Argentieri, R.L.; Farrell, F.X.; Bracht, M.; Sheng, H.; Whitaker, B.; Beck, H.; Tsui, P.; Cochlin, K.; Evanoff, H.L.; et al. Hyper-responsiveness of IPF/UIP fibroblasts: Interplay between TGFbeta1, IL-13 and CCL2. Int. J. Biochem. Cell Biol. 2008, 40, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Okazaki, H.; Sugawara, I.; Yamamoto, K.; Takizawa, H. Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int. Arch. Allergy Immunol. 2003, 132, 168–176. [Google Scholar] [CrossRef]
- Hu, Z.J.; Xu, J.; Yin, J.M.; Li, L.; Hou, W.; Zhang, L.L.; Zhou, Z.; Yu, Y.Z.; Li, H.J.; Feng, Y.M.; et al. Lower Circulating Interferon-Gamma Is a Risk Factor for Lung Fibrosis in COVID-19 Patients. Front. Immunol. 2020, 11, 585647. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bronchial Mucosa Model (bro-ALI) | ||
|---|---|---|
| Canonical Pathway | p Value | Molecules |
| Role of hypercytokinemia/hyperchemokinemia in the pathogenesis of influenza | 2.51 × 10−12 | CXCL10, DDX58, EIF2AK2, IFIT2, IFIT3, IRF7, ISG15, MX1, OAS2, RSAD2 |
| Interferon signaling | 1.29 × 10−10 | IFI6, IFIT1, IFIT3, IFITM1, IFITM2, ISG15, MX1 |
| Role of pattern recognition receptors in recognition of bacteria and viruses | 4.90 × 10−4 | DDX58, EIF2AK2, IFIH1, IRF7, OAS2 |
| Role of RIG1-like receptors in antiviral innate immunity | 9.33 × 10−4 | DDX58, IFIH1, IRF7 |
| WNT/β-catenin signaling | 5.89 × 10−3 | CSNK2A1, MMP7, PPP2CB, TLE4 |
| Sirtuin signaling pathway | 7.41 × 10−3 | H1-3, MAPK7, SCNN1A, TIMM8A, WRN |
| Coronavirus replication pathway | 1.51 × 10−2 | IFITM1, IFITM2 |
| Role of PKR in interferon induction and antiviral response | 1.91 × 10−2 | DDX58, EIF2AK2, IFIH1 |
| Alveolar Mucosa Model (alv-ALI) | ||
| Canonical pathway | p Value | Molecules |
| p53 signaling | 4.07 × 10−3 | BBC3, GNL3, TIGAR |
| APRIL (a proliferation-inducing ligand)-mediated signaling | 8.32 × 10−3 | FOS, TNFSF13 |
| Tight junction signaling | 2.04 × 10−2 | CLDN11, FOS, MYH9 |
| Integrin linked kinase signaling | 2.69 × 10−2 | FOS, MYH9, RSU1 |
| Agranulocyte adhesion, and diapedesis | 3.31 × 10−2 | CLDN11, CXCL14, MYH9 |
| Interleukin-1 signaling | 3.80 × 10−2 | FOS, GNB4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.; Irmler, M.; Keshavan, S.; Introna, M.; Beckers, J.; Palmberg, L.; Johanson, G.; Ganguly, K.; Upadhyay, S. Differential Effect of SARS-CoV-2 Spike Glycoprotein 1 on Human Bronchial and Alveolar Lung Mucosa Models: Implications for Pathogenicity. Viruses 2021, 13, 2537. https://doi.org/10.3390/v13122537
Rahman M, Irmler M, Keshavan S, Introna M, Beckers J, Palmberg L, Johanson G, Ganguly K, Upadhyay S. Differential Effect of SARS-CoV-2 Spike Glycoprotein 1 on Human Bronchial and Alveolar Lung Mucosa Models: Implications for Pathogenicity. Viruses. 2021; 13(12):2537. https://doi.org/10.3390/v13122537
Chicago/Turabian StyleRahman, Mizanur, Martin Irmler, Sandeep Keshavan, Micol Introna, Johannes Beckers, Lena Palmberg, Gunnar Johanson, Koustav Ganguly, and Swapna Upadhyay. 2021. "Differential Effect of SARS-CoV-2 Spike Glycoprotein 1 on Human Bronchial and Alveolar Lung Mucosa Models: Implications for Pathogenicity" Viruses 13, no. 12: 2537. https://doi.org/10.3390/v13122537