Author Contributions
Conceptualization, J.J. and T.C.; methodology, T.C., J.J. and K.-S.S.; software, T.C., J.J. and H.-S.H.; validation, J.J., T.C. and K.-S.S.; formal analysis, T.C. and J.J.; investigation, E.-M.P., T.C. and J.J.; resources, E.-M.P.; data curation, T.C.; writing—original draft preparation, T.C., J.J., K.-S.S. and H.-S.H.; writing—review and editing, T.C., H.-S.H. and K.-S.S.; visualization, J.J., T.C. and H.-S.H.; supervision, T.C. and H.-S.H.; project administration, K.-S.S. and T.C. All authors read and agreed to the published version of the manuscript.
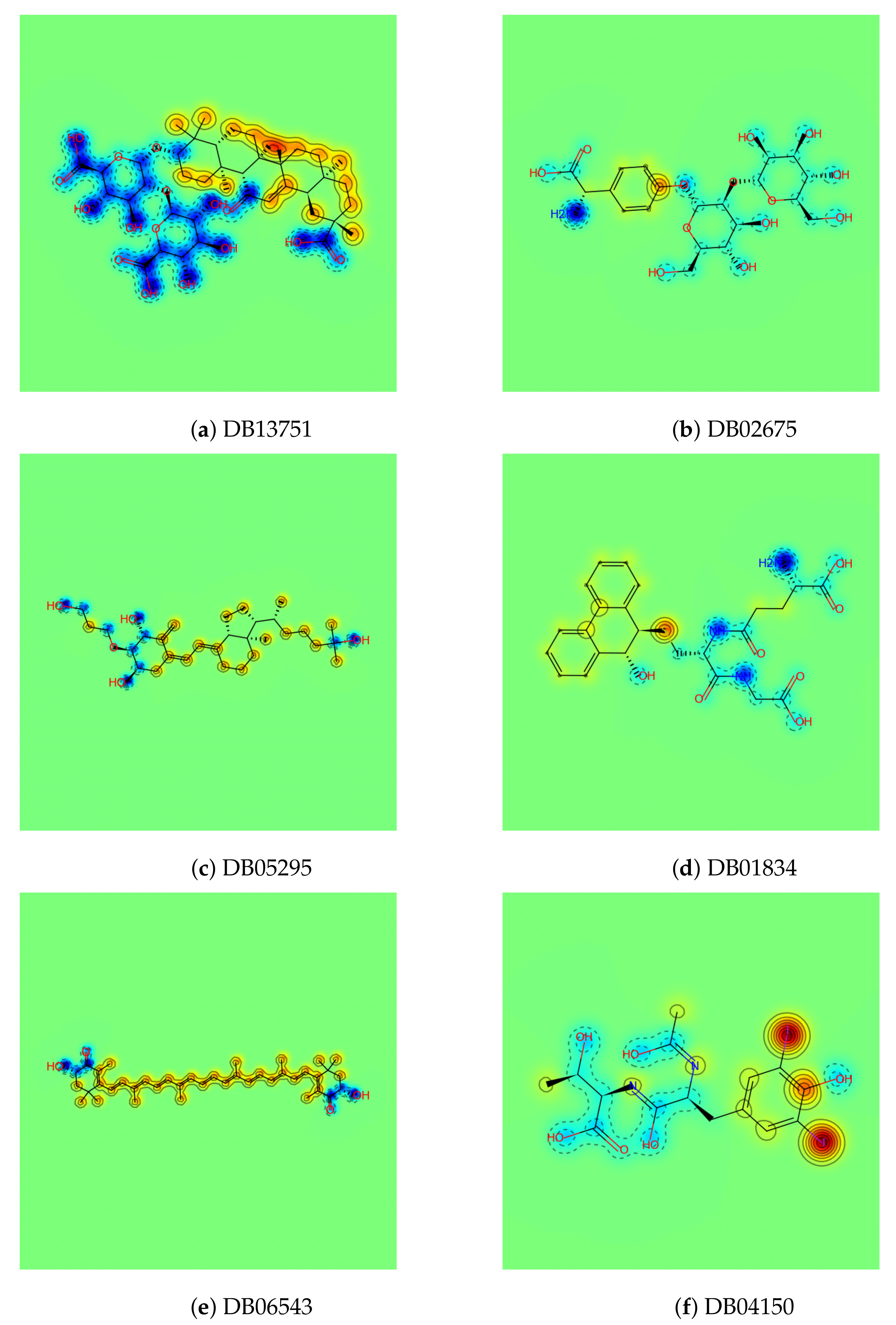
Figure 1.
Six final carrier candidates found by the CSS method and qualitative examination. The color on the similarity maps represents the atom-wise partition coefficient logP: the blue area indicates the hydrophilic atoms having low logP, and the red indicates the hydrophobic atoms having a high logP.
Figure 1.
Six final carrier candidates found by the CSS method and qualitative examination. The color on the similarity maps represents the atom-wise partition coefficient logP: the blue area indicates the hydrophilic atoms having low logP, and the red indicates the hydrophobic atoms having a high logP.
Figure 2.
Result of single layer modeling of DB13751 between water and nafamostat performed by using the Schrödinger software. The result shows that a single layer of DB13751 was formed with some molecules of DB13751 arranged vertically with their hydrophilic heads directed to water (top) and nafamostat (bottom).
Figure 2.
Result of single layer modeling of DB13751 between water and nafamostat performed by using the Schrödinger software. The result shows that a single layer of DB13751 was formed with some molecules of DB13751 arranged vertically with their hydrophilic heads directed to water (top) and nafamostat (bottom).
Figure 3.
TEM images of NM-loaded micelle NPs.
Figure 3.
TEM images of NM-loaded micelle NPs.
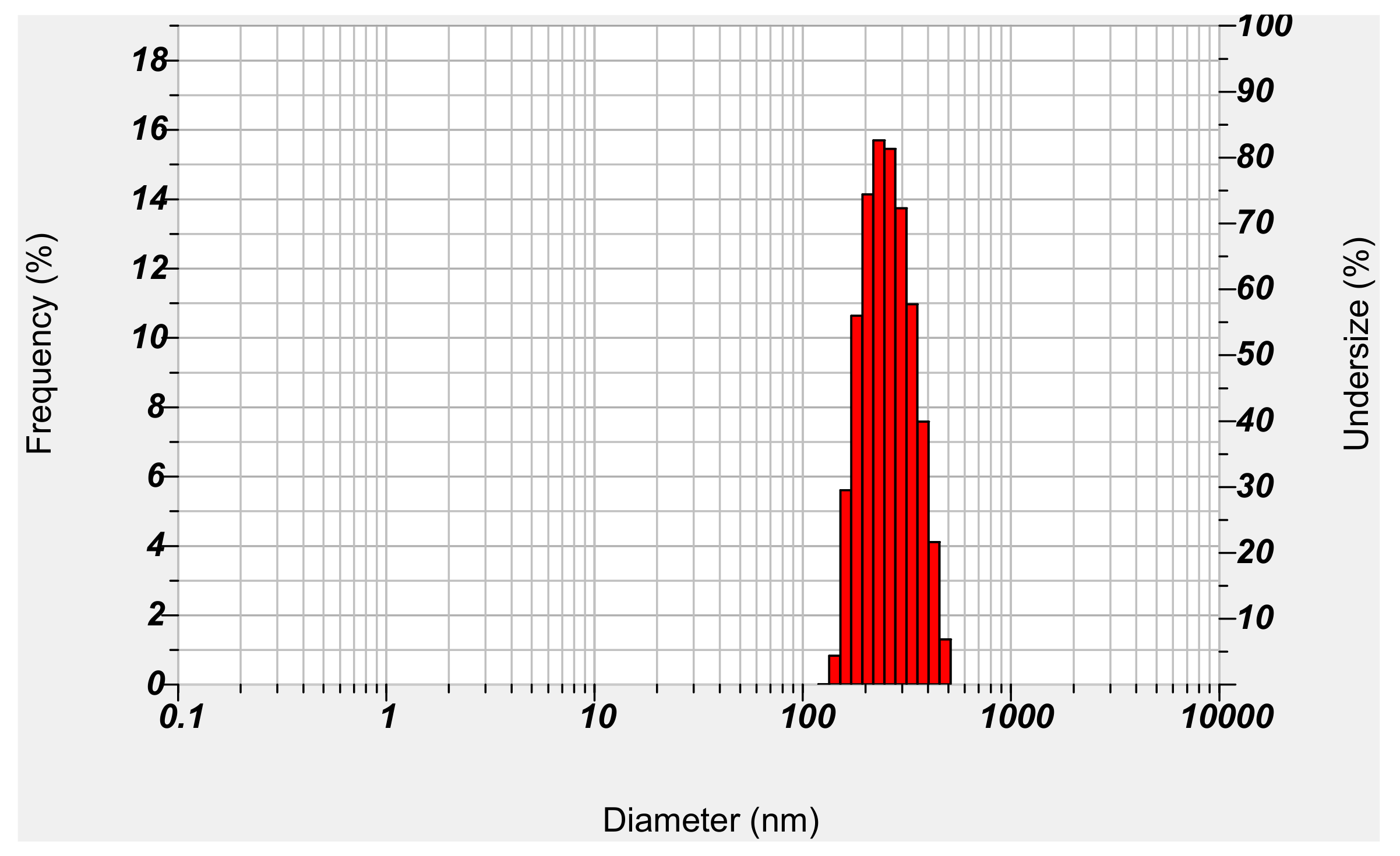
Figure 4.
Particle size and distribution of NM-loaded micelle NPs.
Figure 4.
Particle size and distribution of NM-loaded micelle NPs.
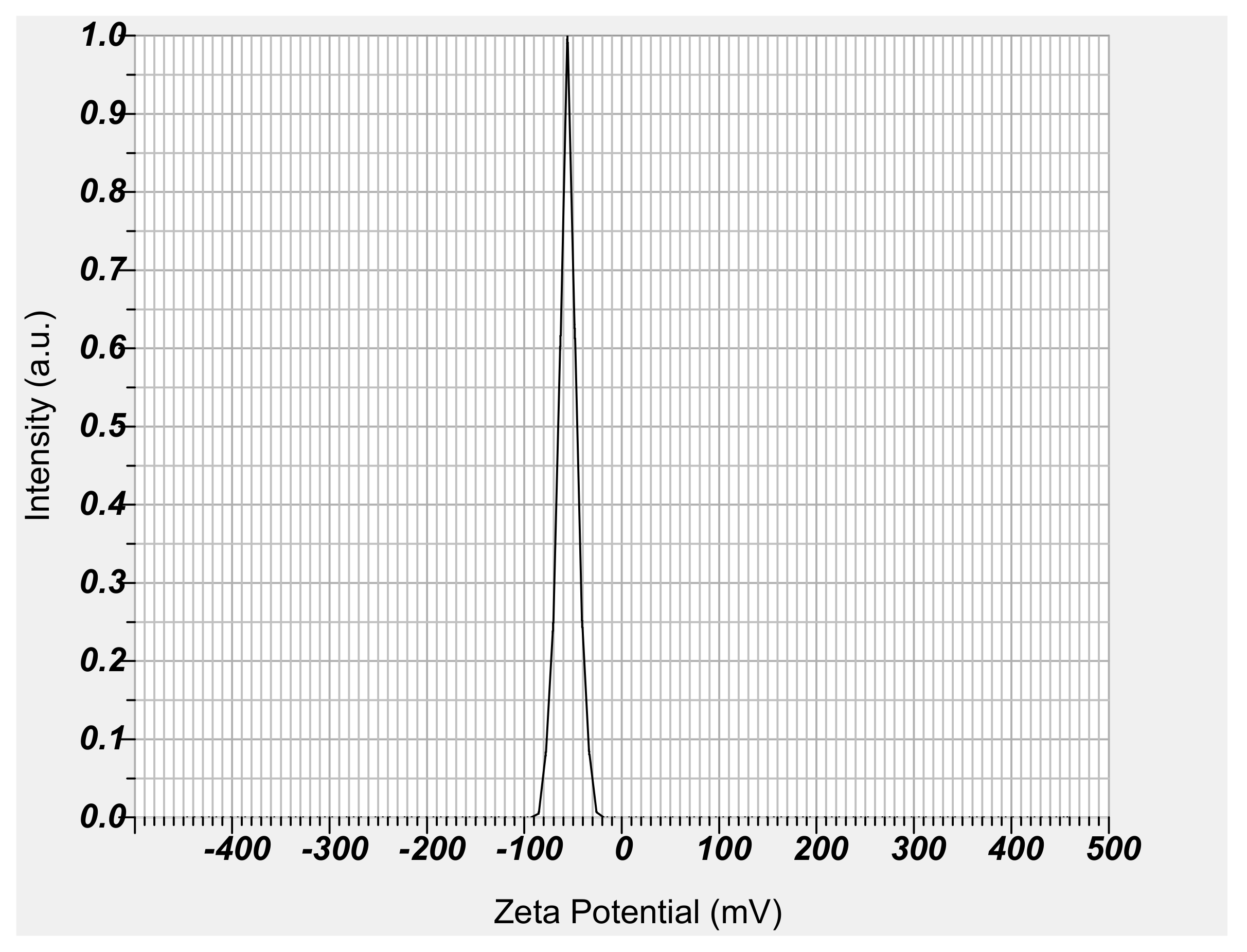
Figure 5.
Zeta potential of NM-loaded micelle NPs.
Figure 5.
Zeta potential of NM-loaded micelle NPs.
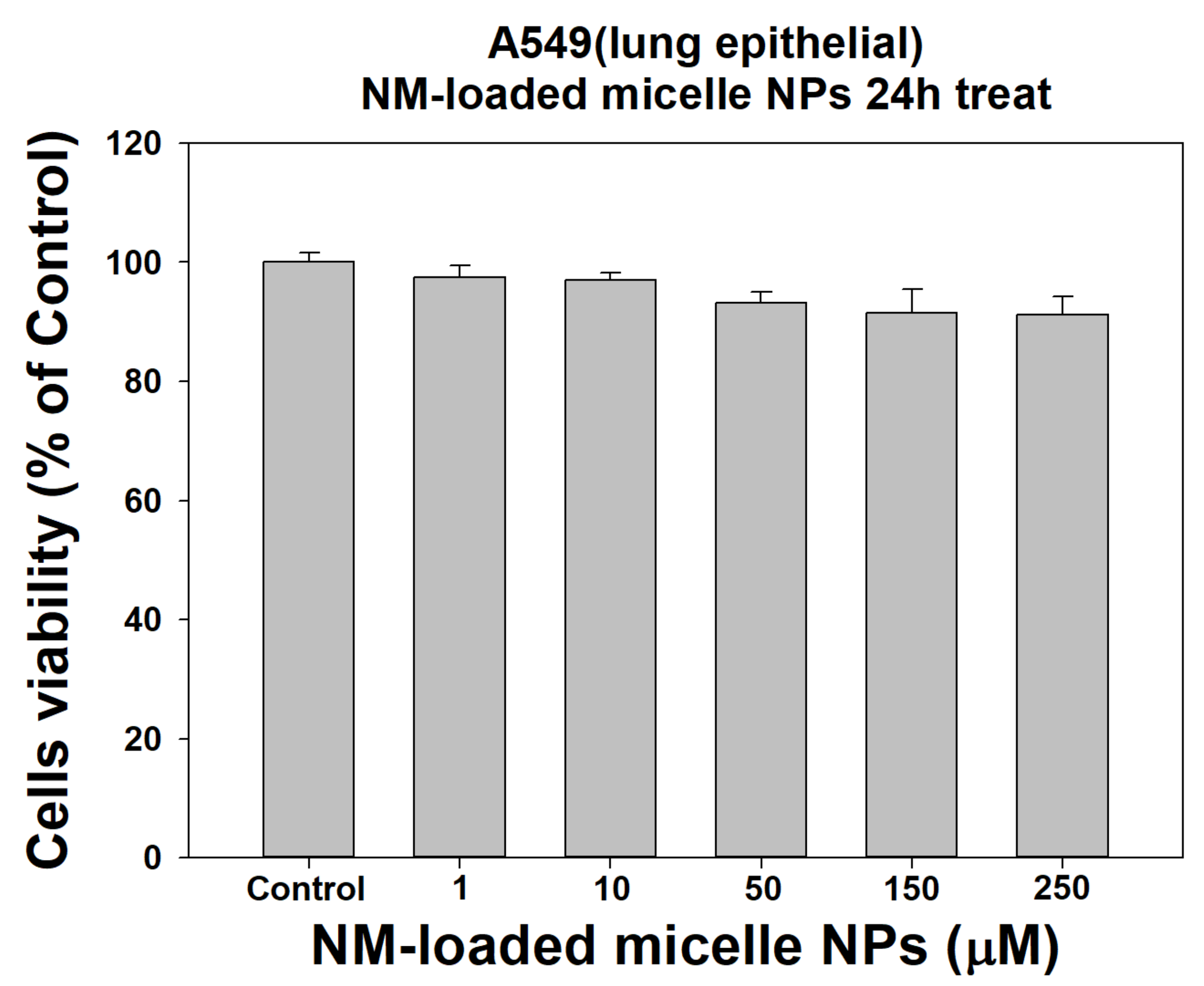
Figure 6.
Cytotoxicity of control and NM-loaded micelle NPs (1, 10, 50, 150, and 250 µM) against A549 cells at one day (n = 8).
Figure 6.
Cytotoxicity of control and NM-loaded micelle NPs (1, 10, 50, 150, and 250 µM) against A549 cells at one day (n = 8).
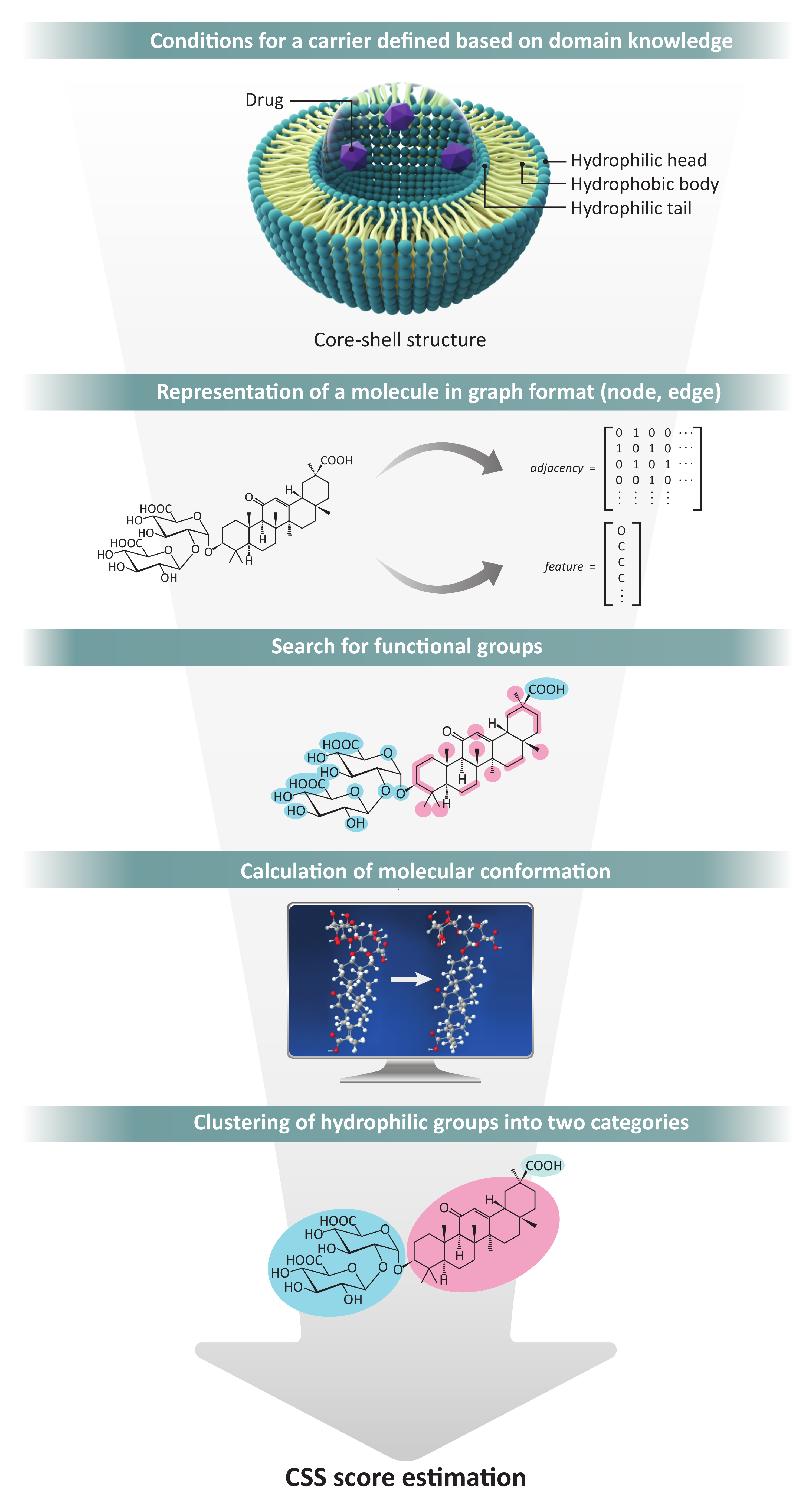
Figure 7.
Total process of the CSS method.
Figure 7.
Total process of the CSS method.
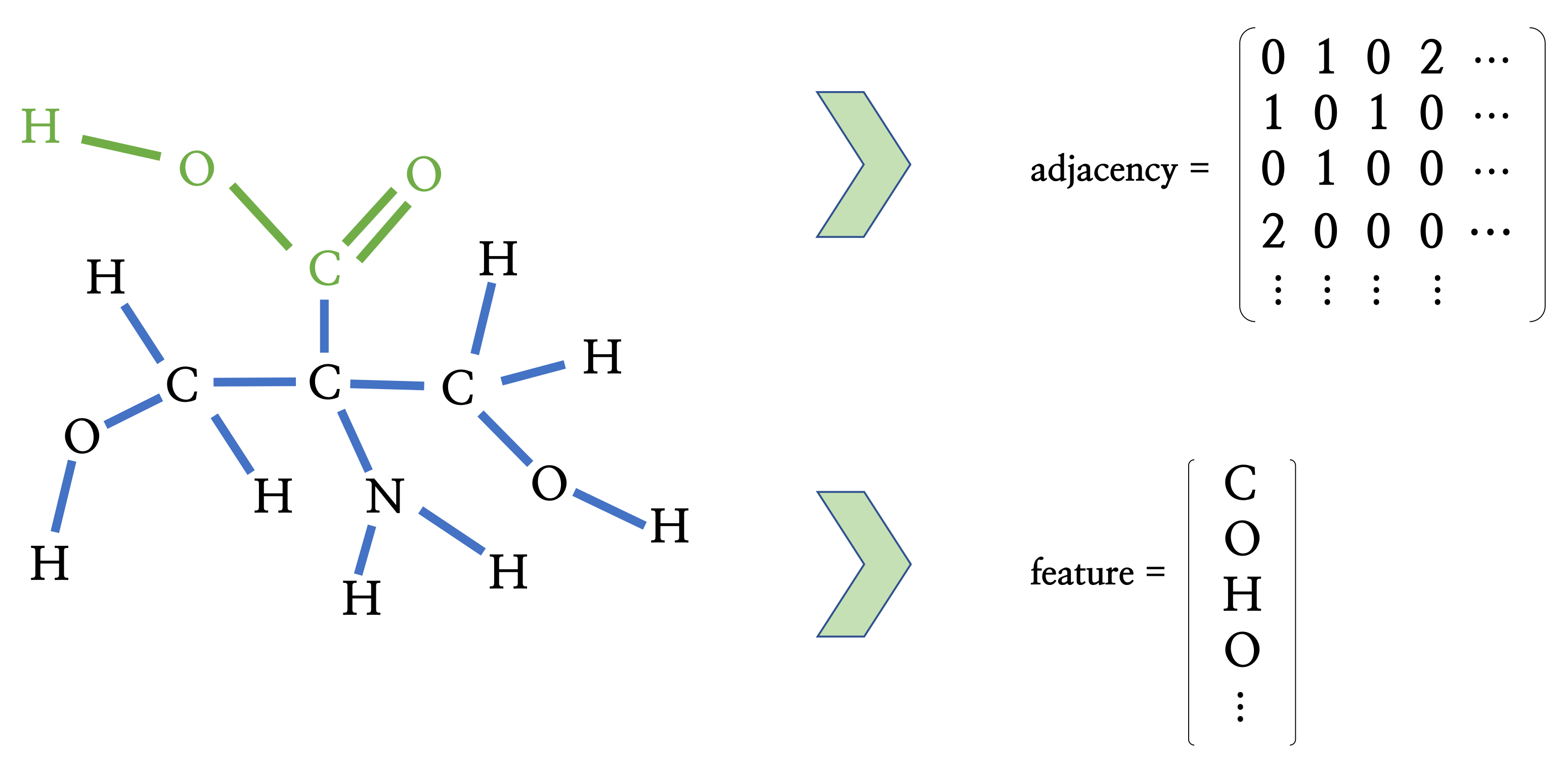
Figure 8.
The example of the process representing molecules in graph format. When a molecule has N atoms, elements of the adjacency matrix denote the bond type between atomic pairs and elements of the feature vector , which denotes the atomic numbers. We can see that a carboxylic group (marked in green) is represented in the adjacency matrix and feature vector.
Figure 8.
The example of the process representing molecules in graph format. When a molecule has N atoms, elements of the adjacency matrix denote the bond type between atomic pairs and elements of the feature vector , which denotes the atomic numbers. We can see that a carboxylic group (marked in green) is represented in the adjacency matrix and feature vector.
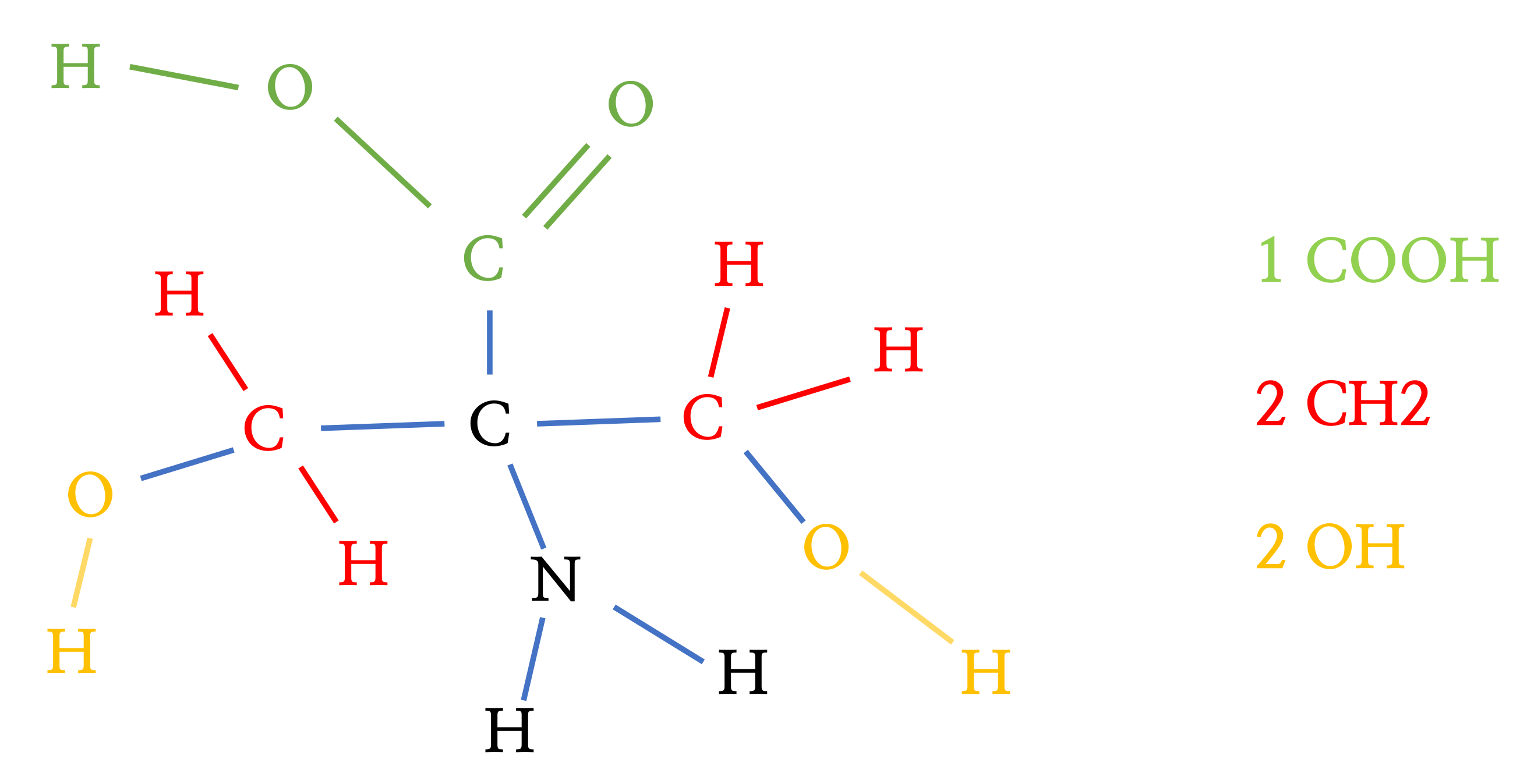
Figure 9.
Subtracted groups of molecule.
Figure 9.
Subtracted groups of molecule.
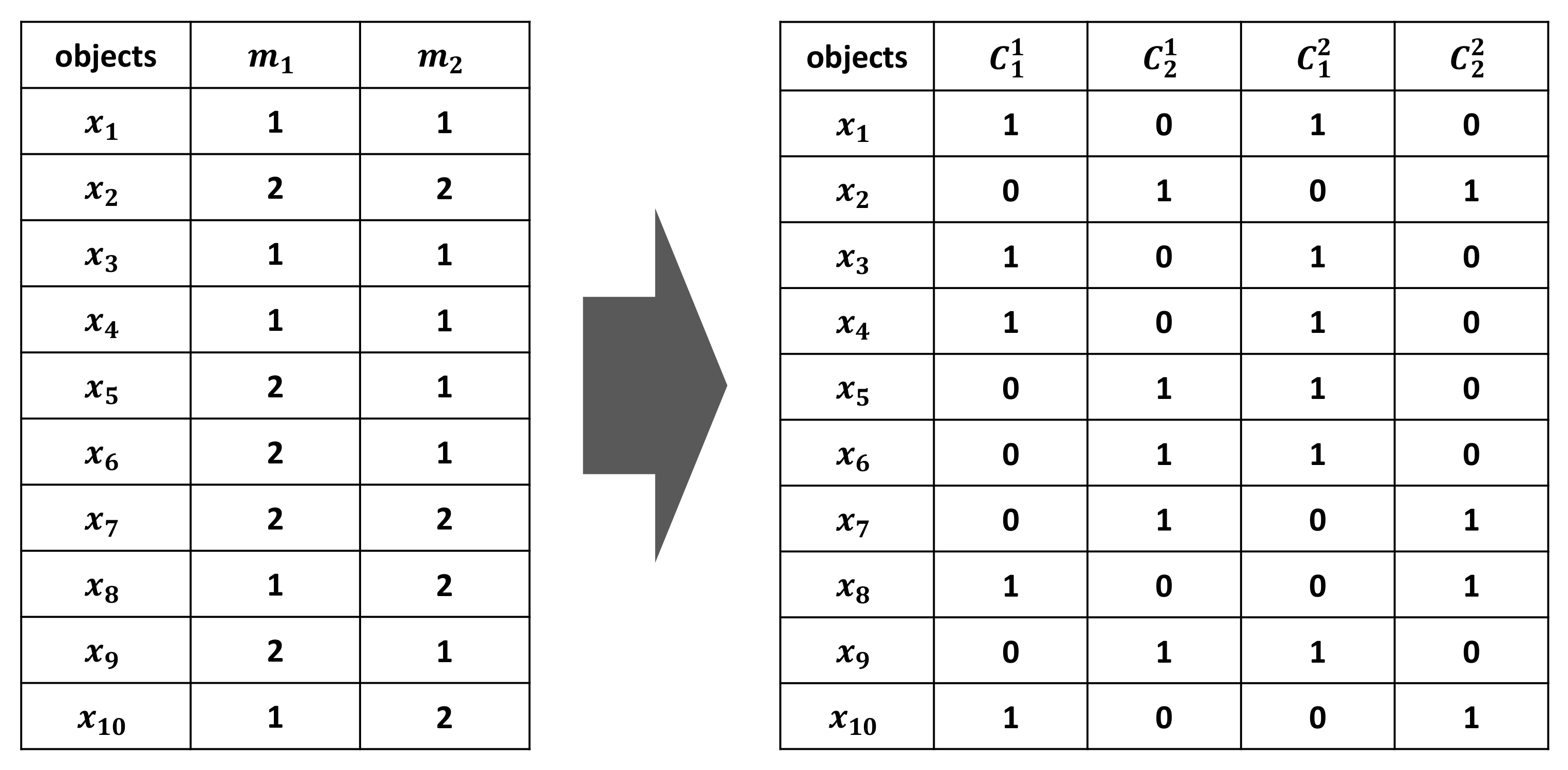
Figure 10.
An illustrative example of two clustering members for a dataset with 10 objects.
Figure 10.
An illustrative example of two clustering members for a dataset with 10 objects.
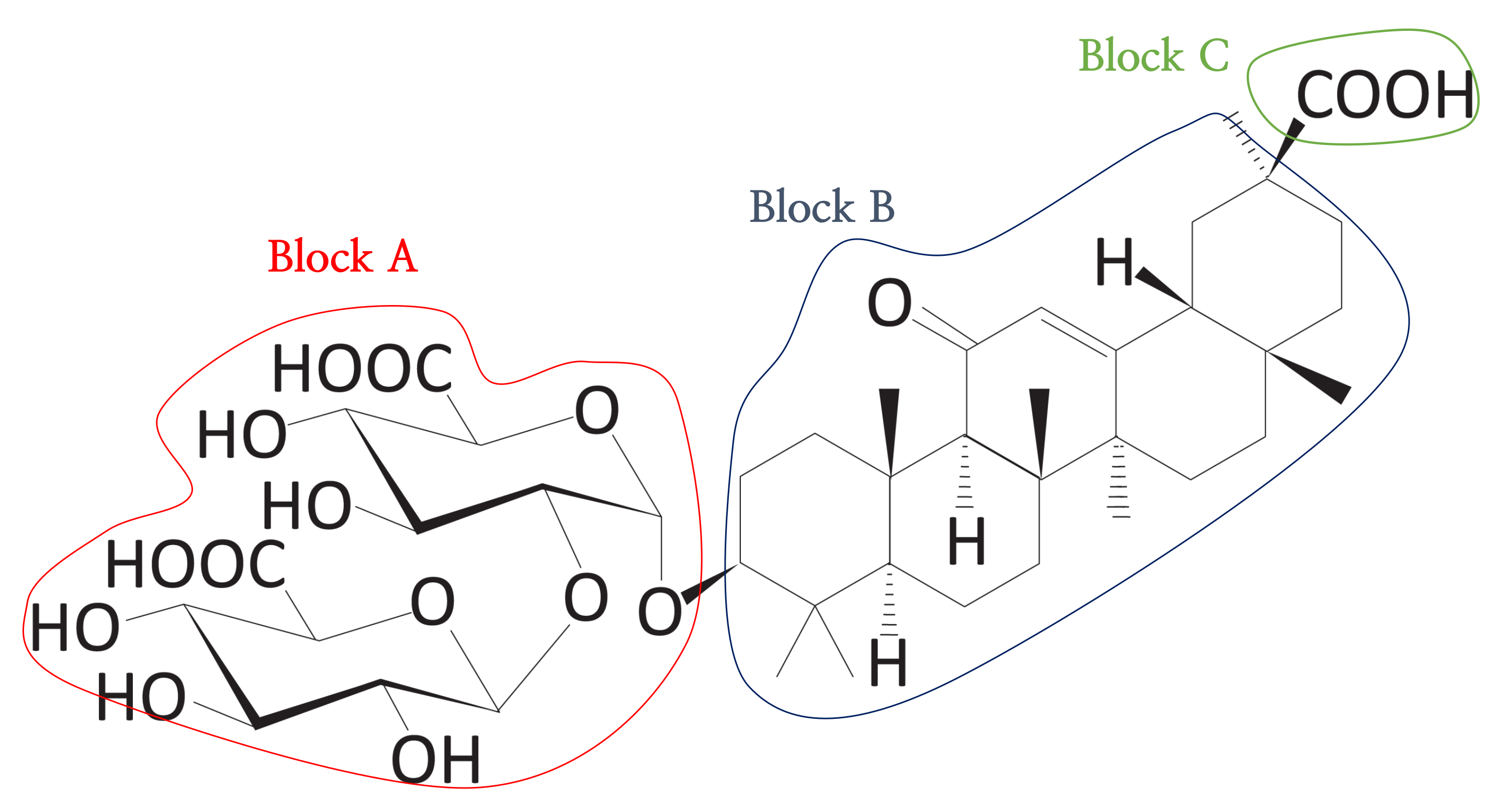
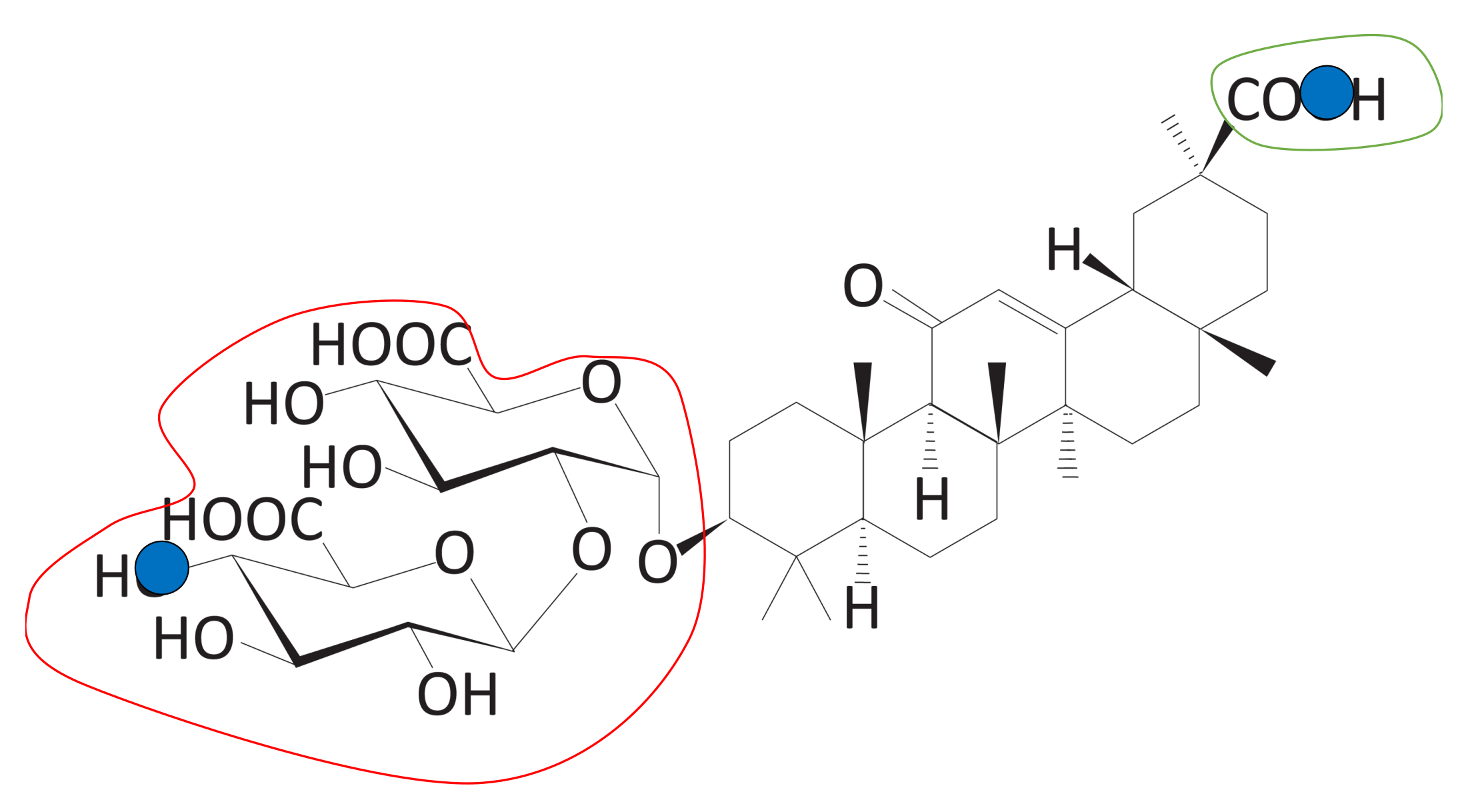
Figure 11.
The triblock form of DB13751. The red and green line areas represent the hydrophilic blocks, and the blue line area represents the hydrophobic group derived from the clustering process. We call the red line area Block A, the blue line area Block B, and the green line area Block C. These blocks are used to calculate the sub-scores.
Figure 11.
The triblock form of DB13751. The red and green line areas represent the hydrophilic blocks, and the blue line area represents the hydrophobic group derived from the clustering process. We call the red line area Block A, the blue line area Block B, and the green line area Block C. These blocks are used to calculate the sub-scores.
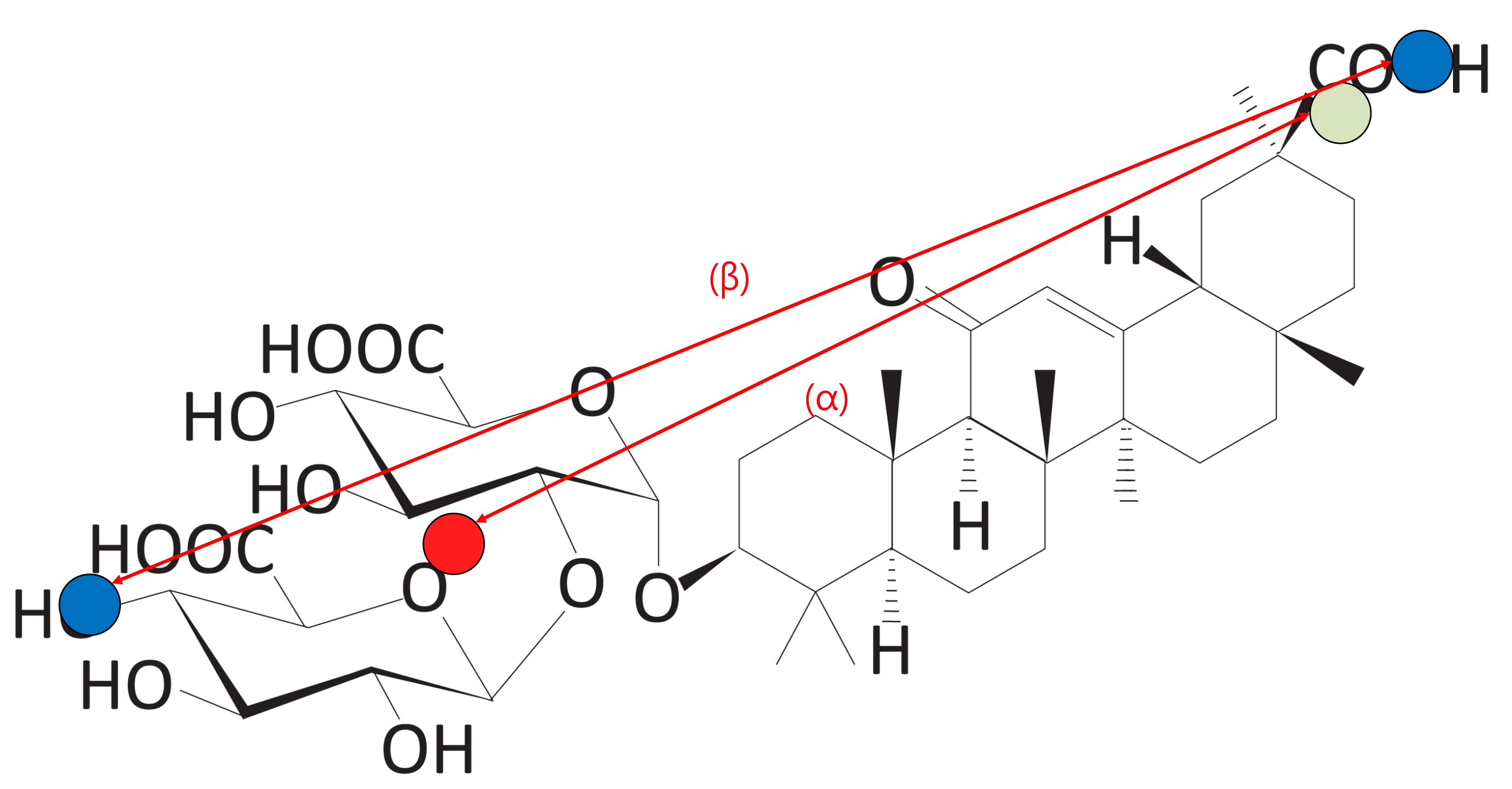
Figure 12.
The distance between the hydrophilic clusters (Score 1). Blue dots represent length of the molecule. The red dot represents the center point of Block A. The green dot represents the center point of Block C.
Figure 12.
The distance between the hydrophilic clusters (Score 1). Blue dots represent length of the molecule. The red dot represents the center point of Block A. The green dot represents the center point of Block C.
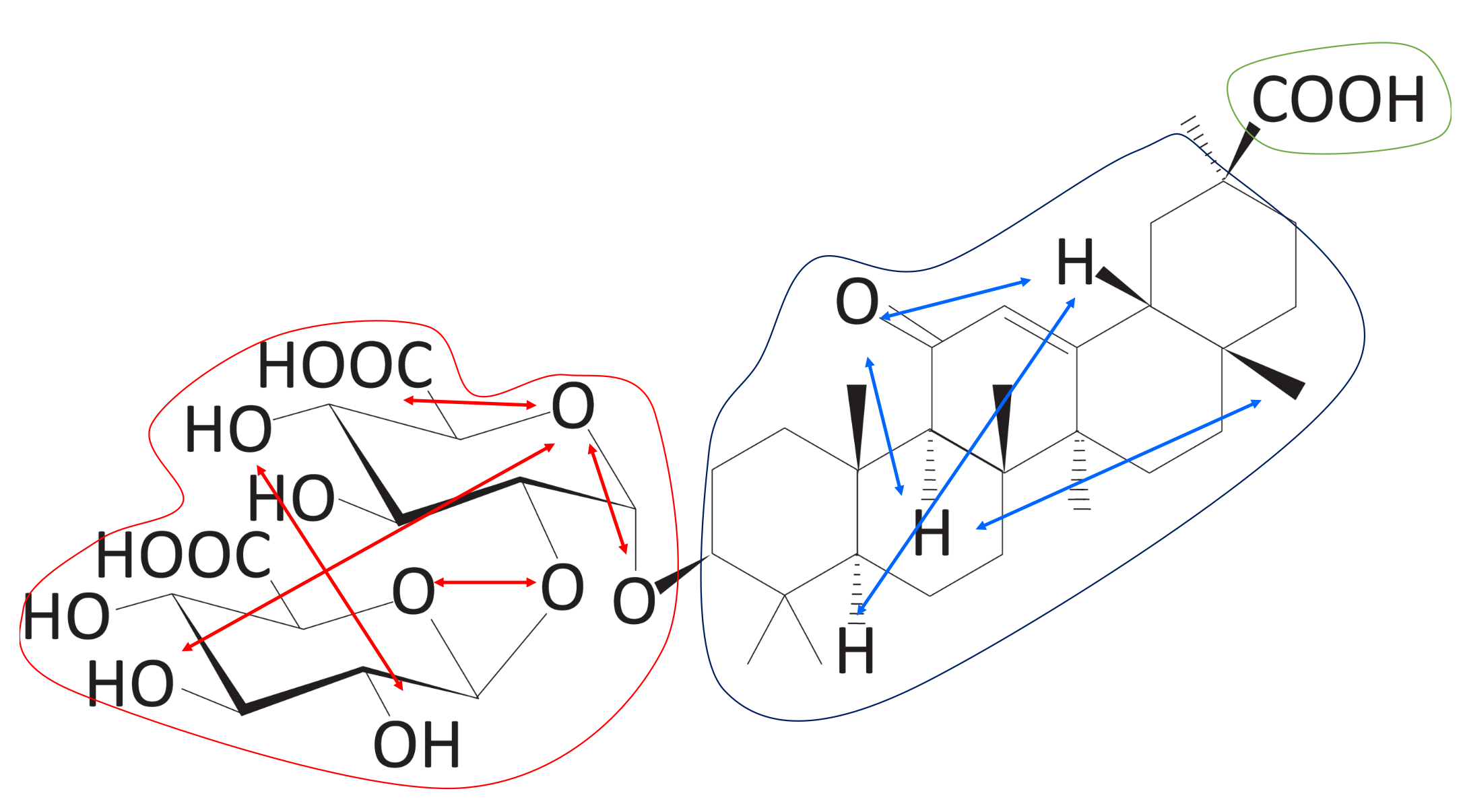
Figure 13.
Within-block distance (Score 2). Red arrows represent the distance between groups belonging to Block A. Blue arrows represent the distance between groups belonging to Block B. In this case, Block C has only one group. These distances were used to measure Score 2 (Algorithm 3).
Figure 13.
Within-block distance (Score 2). Red arrows represent the distance between groups belonging to Block A. Blue arrows represent the distance between groups belonging to Block B. In this case, Block C has only one group. These distances were used to measure Score 2 (Algorithm 3).
Figure 14.
Size asymmetry between Block A and Block C (Score 3). The number of groups belonging to Blocks A and Block C can be compared.
Figure 14.
Size asymmetry between Block A and Block C (Score 3). The number of groups belonging to Blocks A and Block C can be compared.
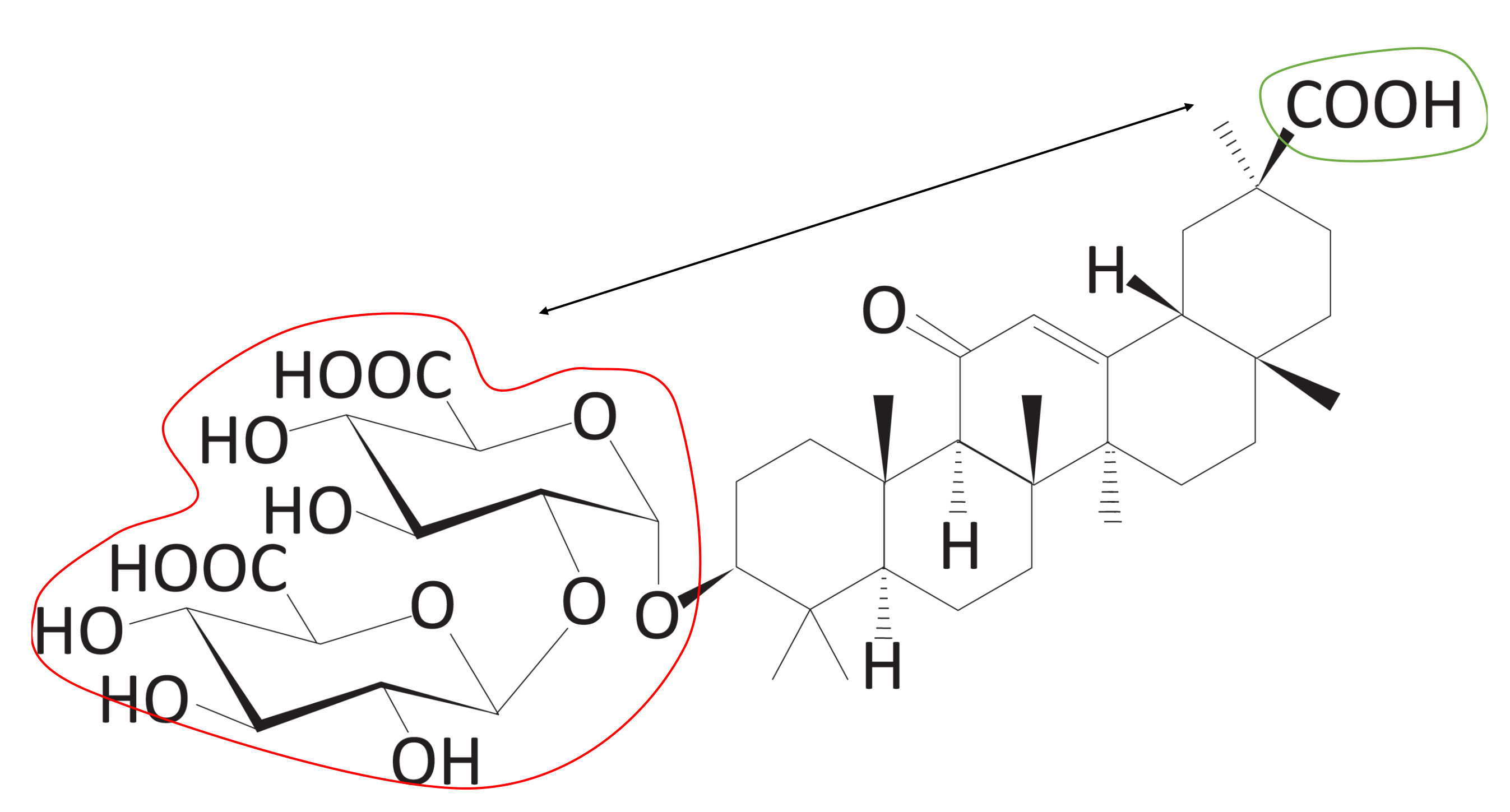
Figure 15.
Number of head and tail atoms included in the hydrophilic group (Score 4). The blue dots represent the head and tail atoms, which means two atoms that have the longest distance within a molecule. Head and tail atoms can be checked to determine whether they belong to hydrophilic groups.
Figure 15.
Number of head and tail atoms included in the hydrophilic group (Score 4). The blue dots represent the head and tail atoms, which means two atoms that have the longest distance within a molecule. Head and tail atoms can be checked to determine whether they belong to hydrophilic groups.
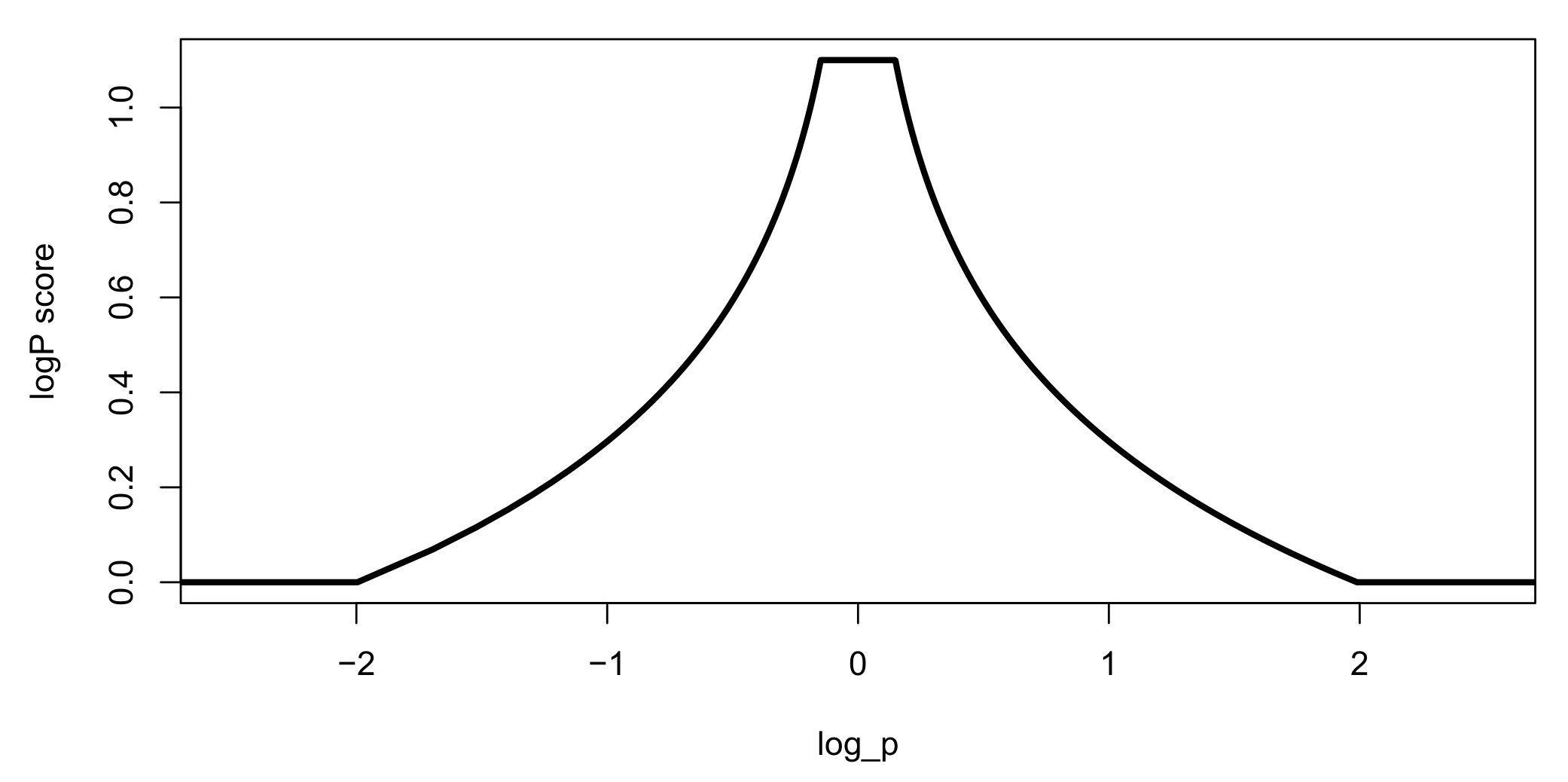
Figure 16.
LogP score. The closer the logP value is to zero, the higher the score. The maximum value of the score is 1.1, and the minimum value is zero.
Figure 16.
LogP score. The closer the logP value is to zero, the higher the score. The maximum value of the score is 1.1, and the minimum value is zero.
Table 1.
CS scores of the molecules after qualitative examination. The rows are drugs listed in the DrugBank 5.0 database. The columns show sub-scores that constitute the CS score. Scores 1–4 mean the distance between Block A and C, within-block distance, size asymmetry between Block A and Block C, and number of head and tail atoms included in the hydrophilic group, respectively. The logP score is not the value of logP, and the largest value was regularized when the value of logP was 0. The following sentence indicates the official name of the DB identifier. DB13751:glycyrrhizic acid; DB02675:(4-hydroxymaltosephenyl)glycine; DB05295:eldecalcitol; DB01834:(9R,10R)-9-(S-glutathionyl)-10-hydroxy-9,10-dihydrophenanthrene; DB06543:astaxanthin; DB04150:threonine derivative.
Table 1.
CS scores of the molecules after qualitative examination. The rows are drugs listed in the DrugBank 5.0 database. The columns show sub-scores that constitute the CS score. Scores 1–4 mean the distance between Block A and C, within-block distance, size asymmetry between Block A and Block C, and number of head and tail atoms included in the hydrophilic group, respectively. The logP score is not the value of logP, and the largest value was regularized when the value of logP was 0. The following sentence indicates the official name of the DB identifier. DB13751:glycyrrhizic acid; DB02675:(4-hydroxymaltosephenyl)glycine; DB05295:eldecalcitol; DB01834:(9R,10R)-9-(S-glutathionyl)-10-hydroxy-9,10-dihydrophenanthrene; DB06543:astaxanthin; DB04150:threonine derivative.
| Rank | DB ID | Score 1 | Score 2 | Score 3 | Score 4 | logP Score | Total Score |
|---|
| | | | | ⋮ | | | |
| 3 | DB13751 | 0.725 | 0.651 | 11 | 2 | 0 | 14.403 |
| | | | | ⋮ | | | |
| 18 | DB02675 | 0.646 | 0.703 | 5.5 | 2 | 0 | 10.118 |
| | | | | ⋮ | | | |
| 35 | DB05295 | 0.737 | 0.746 | 4 | 2 | 0 | 9.332 |
| | | | | ⋮ | | | |
| 74 | DB01834 | 0.582 | 0.643 | 5 | 1 | 0.632 | 8.09 |
| | | | | ⋮ | | | |
| 82 | DB06543 | 0.997 | 0.625 | 1 | 2 | 0 | 8.057 |
| | | | | ⋮ | | | |
| 124 | DB04150 | 0.6829 | 0.65 | 4 | 1 | 0 | 7.524 |
| | | | | ⋮ | | | |
Table 2.
Hydrophilic-hydrophobic group.
Table 2.
Hydrophilic-hydrophobic group.
| Hydrophilic Groups | Hydrophobic Groups |
|---|
| –NR(tertiary amine) | –CH– |
| –COO(sorbitan ring) | –CH– |
| –COO | –CH– |
| –COOH | |
| –OH | |
| –O– | |
| –OH(sorbitan ring) | |
| –CHCHO– | |




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}