
Abstract
An ideal therapeutic anti-SARS-CoV-2 antibody would resist viral escape1,2,3, have activity against diverse sarbecoviruses4,5,6,7, and be highly protective through viral neutralization8,9,10,11 and effector functions12,13. Understanding how these properties relate to each other and vary across epitopes would aid the development of therapeutic antibodies and guide vaccine design. Here we comprehensively characterize escape, breadth and potency across a panel of SARS-CoV-2 antibodies targeting the receptor-binding domain (RBD). Despite a trade-off between in vitro neutralization potency and breadth of sarbecovirus binding, we identify neutralizing antibodies with exceptional sarbecovirus breadth and a corresponding resistance to SARS-CoV-2 escape. One of these antibodies, S2H97, binds with high affinity across all sarbecovirus clades to a cryptic epitope and prophylactically protects hamsters from viral challenge. Antibodies that target the angiotensin-converting enzyme 2 (ACE2) receptor-binding motif (RBM) typically have poor breadth and are readily escaped by mutations despite high neutralization potency. Nevertheless, we also characterize a potent RBM antibody (S2E128) with breadth across sarbecoviruses related to SARS-CoV-2 and a high barrier to viral escape. These data highlight principles underlying variation in escape, breadth and potency among antibodies that target the RBD, and identify epitopes and features to prioritize for therapeutic development against the current and potential future pandemics.
Similar content being viewed by others


Main
The most potently neutralizing antibodies to SARS-CoV-2—including those in clinical use14 and dominant in polyclonal sera15,16—target the spike RBD. Mutations in the RBD that reduce binding by antibodies have emerged among SARS-CoV-2 variants17,18,19,20,21, highlighting the need for antibodies and vaccines that are robust to viral escape. We have previously described S3094, an antibody that exhibits potent effector functions and neutralizes all current SARS-CoV-2 variants22,23 and the divergent sarbecovirus SARS-CoV-1. S309 forms the basis for an antibody therapy (VIR-7831, recently renamed sotrovimab) that has received emergency use authorization from the US Food and Drug Administration for treatment of COVID-1924. Longer term, antibodies with broad activity across sarbecoviruses would be useful to combat potential future spillovers6. These efforts would be aided by a systematic understanding of the relationships between antibody epitope, resistance to viral escape, and breadth of sarbecovirus cross-reactivity. Here we address this question by comprehensively characterizing a diverse panel of antibodies, including S309, using deep mutational scanning, pan-sarbecovirus binding assays, in vitro selection of viral escape, and biochemical and structural analyses.
Antibody potency, escapability and breadth
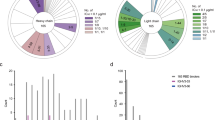
We identified a panel of anti-SARS-CoV-2 antibodies with distinct properties (Fig. 1a, Extended Data Table 1), including six antibodies newly described in this study. These antibodies bind different epitopes within the RBM and the non-RBM ‘core’ of the RBD. The antibody panel spans a range of neutralization potencies and binding affinities (Fig. 1a, Extended Data Fig. 1a–c).

a, SARS-CoV-2 neutralization potency (authentic virus (n = 3) and spike-pseudotyped VSV particles (VSV pp) (n = 3 to 8) on Vero E6 cells), Fab–RBD binding affinities measured by surface plasmon resonance (SPR) (n = 2 to 4), and epitope classifications. Additional details in Extended Data Table 1. Asterisks indicate antibodies that are newly described in this paper. Epitope classes are described in refs. 9,15. IC50, half-maximal inhibitory concentration; KD, dissociation constant; NA, not applicable. b, c, Left, deep mutational scanning maps of mutations that escape binding by antibodies targeting the core RBD (b) or the RBM (c). Letter height indicates the strength of the mutation’s escape from antibody binding. Letters coloured by effect on folded RBD expression (b) or ACE2 binding affinity26 (c). Right, relative functional epitope size and escapability, scaling from 0 (no escape mutations) to 1 (largest epitope and most escapable antibody). Bottom, heat maps illustrate variability among sarbecoviruses or SARS-CoV-2 sequences. d, Antibody binding to a pan-sarbecovirus RBD panel. Heat map illustrates binding from fluorescence-activated cell sorting (FACS)-based selections (colour bar, bottom left). Asterisks indicate reduced-affinity binding in secondary binding assays (Extended Data Fig. 4a–f).
We used deep mutational scanning to map how all amino-acid mutations in the SARS-CoV-2 RBD affect binding by each antibody3 (Fig. 1b, c, Extended Data Fig. 2). Some antibodies have narrowly focused functional epitopes (the set of residues in which mutations abolish binding25), with binding-escape mutations at just a few key residues (for example, S309, S2D106), whereas others have wider functional epitopes (for example, S2H13; Fig. 1b, c). We previously measured how all RBD mutations affect folded RBD expression and ACE2 binding affinity26 (Fig. 1b, c, letter colours). We used the combined measures of how mutations affect antibody binding and RBD function to compute the ‘escapability’ of each antibody, which reflects the extent to which mutations that escape antibody binding are functionally tolerated (Fig. 1b, c, Extended Data Fig. 3a, b). We also investigated the sensitivity of each antibody to mutations among SARS-CoV-2 sequences reported in GISAID (Fig. 1b, c, heat map, Extended Data Fig. 3c), and found that some antibodies are more affected by natural SARS-CoV-2 mutations than others, including mutations found in SARS-CoV-2 variants of concern27,28,29 (Extended Data Fig. 1d).
We next extended our deep mutational scanning platform to measure binding of each antibody to a pan-sarbecovirus panel of 45 RBDs (Fig. 1d, Extended Data Fig. 4a–f). The four antibodies that bind the core RBD exhibit cross-reactive binding to RBDs from SARS-CoV-1 and related ACE2-dependent bat sarbecoviruses, and from sarbecoviruses isolated in Europe and Africa. S304 and S2H97 antibodies also bind RBDs of the most divergent clade from Asia, which has an average of 64% amino acid identity with SARS-CoV-2. S2H97 exhibits notably tight binding to all RBDs tested (Fig. 1d, Extended Data Fig. 4f), making it, to our knowledge, the broadest pan-sarbecovirus RBD antibody described to date. Antibodies that bind epitopes within the RBM exhibit more limited cross-reactivity, typically binding only SARS-CoV-2 and the closely related GD-pangolin-CoV RBD. S2E12 stands out among the RBM antibodies we evaluated, as it also binds the RaTG13 and GX-pangolin-CoV RBDs, showing that even within the evolutionarily plastic RBM19,26 there are epitopes that enable greater breadth than others.
The pan-sarbecovirus S2H97 antibody
To understand the structural basis for cross-reactive sarbecovirus binding, we determined the structures of the S2H97 antigen-binding fragment (Fab) (X-ray crystallography, 2.65 Å resolution), S2X35 Fab (X-ray crystallography, 1.83 Å resolution) and S2E12 Fab (X-ray crystallography, 2.95 Å resolution) bound to SARS-CoV-2 RBD (Fig. 2a, Extended Data Table 2). This panel of cross-reactive antibodies emphasizes the core RBD as a general target of broad antibody binding owing to its conservation among sarbecoviruses, reflected in the diverse core RBD surfaces targeted by the broadest of these antibodies (Fig. 2a, Extended Data Fig. 5a–g).

a, Composite model of the SARS-CoV-2 trimer with cross-reactive antibodies. Epitopes recognized by each Fab are shown as coloured surface and ACE2 footprint is shown as a black outline. b, Integrative features of the S2H97 structural footprint (5 Å cut-off). Heat maps illustrate evolutionary variability (blue), functional constraint from previous deep mutational scans (grey) and energetic contribution to binding from the static crystal structure (predicted energy) or molecular dynamics (MD) simulation (green). Logo plot as in Fig. 1b. Asterisk, introduction of N-linked glycosylation motifs. c, S2H97 breadth of neutralization (spike-pseudotyped VSV on Vero E6 cells). Curves representative of at least two independent experiments. Data are mean ± s.d. from three technical replicates. Geometric mean IC50 is shown. d, S2H97 neutralization of SARS-CoV-2 variants (Extended Data Fig. 1d; spike-pseudotyped VSV on Vero E6 cells). Points represent individual measurements and bars show mean fold-change in neutralization potency relative to the Wuhan-1 strain with the D614G S mutation. e, Negative-stain electron microscopy imaging of native-like soluble prefusion S trimer alone (left) or incubated with S2H97 (right). Micrographs are representative of 51 (SARS-CoV-2 S) and 173 (S + S2H97) micrographs. f, S2H97 prophylactic efficacy in Syrian hamsters. Median infectious virus titres (left, TCID50) and RNA levels (right) 4 days after SARS-CoV-2 challenge in hamsters prophylactically dosed with 25 mg kg−1 S2H97 or isotype control. The double daggers indicate two hamsters with undetectable S2H97 levels (below 50 ng ml−1) at the time of viral challenge. **P = 0.0048 (virus titre) and P = 0.0048 (RNA) vs control, two-sided Mann–Whitney test (hamsters with no detectable serum antibody excluded). g, Blockade of binding15 by sera from SARS-CoV-2-infected (top) or vaccinated (bottom) individuals. BD80 is the concentration that blocks 80% of binding. Percentages of samples with blockade above the lower detection limit are indicated. WT, wild type.
The exceptionally cross-reactive S2H97 antibody targets a previously undescribed cryptic antigenic site, which we designated site V (Fig. 2a, b). S2H97 binding is facilitated by packing of the heavy chain complementarity-determining region 3 (CDR3) into an RBD crevice at the centre of the epitope, together with polar contacts with all three heavy chain CDRs and the light chain CDR2 (Extended Data Fig. 5f). Molecular dynamics simulation of the S2H97 Fab–RBD complex highlights the durability of many of these interactions (Fig. 2b). The surface bound by S2H97 is constrained by the deleterious effects of mutations on folded RBD expression26 (Fig. 2b), and this constraint is probably enhanced by quaternary packing with the N-terminal domain in the closed spike trimer (Extended Data Fig. 6a). Consistent with the conservation of the S2H97 epitope, S2H97 neutralizes diverse sarbecoviruses (Fig. 2c, Extended Data Fig. 4g) and SARS-CoV-2 variants (Fig. 2d).
To understand the evolution of S2H97 breadth, we measured breadth of binding by its germline form, S2H97GL, in which we reverted the 13 somatic mutations (Extended Data Fig. 4h, i). S2H97GL bound all tested sarbecovirus RBDs and exhibited particularly high affinity for SARS-CoV-2-related RBDs. Somatic mutations enhanced affinity across all sarbecoviruses by two orders of magnitude. This general increase in affinity together with the conservative amino acid replacements among paratope residues suggests that framework mutations may contribute to a general improvement in S2H97 binding affinity.
To characterize the mechanism of S2H97 neutralization, we determined a cryo-electron microscopy structure of S2H97 bound to SARS-CoV-2 S (Extended Data Fig. 5i–l, Extended Data Table 3). S2H97 binding requires extensive opening of the RBD to unmask its cognate epitope (Extended Data Fig. 6b), even more than is required to access the cryptic antigenic site II15. Similar to other antibodies that only bind the open RBD30,31, S2H97 induces rapid and premature refolding of spike into the post-fusion state (Fig. 2e), promotes S1 shedding of cell-surface-expressed spike (Extended Data Fig. 6c), and induces a low level of syncytia formation among spike-expressing cells (Extended Data Fig. 6d). S2H97 does not interfere with ACE2 binding (Extended Data Fig. 6e). Like other non-ACE2-competitive antibodies31,32, S2H97 neutralization is attenuated in cells that overexpress ACE2 (Extended Data Fig. 6f). Consistent with its ability to neutralize spike-mediated viral entry, S2H97 inhibits spike-mediated cell–cell fusion (Extended Data Fig. 6g). Together, these experiments suggest that the mechanism of neutralization by S2H97 involves receptor-independent conversion of S to the post-fusion state30, thereby inhibiting ACE2-mediated cell entry.
Next, we determined the prophylactic efficacy of S2H97 in vivo using a Syrian hamster model of infection. We administered hamsters with 25 mg kg−1 S2H97 2 days before intranasal challenge with SARS-CoV-2 and assessed viral RNA load and infectious viral titres in the lungs 4 days after infection. S2H97 prophylaxis reduced the number of RNA copies by more than 10,000-fold relative to control in the four hamsters that had detectable circulating antibody levels at the time of challenge, and reduced infectious viral titres to the lower detection limit in these hamsters (Fig. 2f). The two hamsters not exhibiting a reduction in viral load had circulating S2H97 levels below the limit of quantification (50 ng ml−1) at the time of viral challenge (Extended Data Fig. 6h), which may reflect a failure in the intraperitoneal administration procedure. Therefore, S2H97 demonstrates that antibodies to the newly identified antigenic site V can be protective in vivo.
Finally, we performed serum blockade of binding experiments15, demonstrating that antibodies competing with S2H97 binding are rare in infection- and vaccine-elicited sera (Fig. 2g). This sub-dominance of antigenic site V may be explained by the inaccessibility of the epitope as illustrated in the cryoEM structure. However, the protective nature and exceptional breadth of S2H97 suggests that updated immunogen designs, such as those based on the RBD33,34,35, could unmask antigenic site V to better elicit S2H97-like antibodies.
Breadth and escapability among RBM epitopes
Our survey reveals variation in the escapability and breadth of antibodies that target the RBM (Fig. 1c, d), which is immunodominant (Fig. 2g) but variable over sarbecovirus and SARS-CoV-2 evolution. We performed in vitro selection experiments to identify spike-expressing vesicular stomatitis virus (VSV) mutants that emerge in the presence of each of seven monoclonal antibodies (Fig. 3a, Extended Data Fig. 7a, b) to further understand escape from these antibodies.

a, Escape mutations in spike-expressing VSV passaged in the presence of monoclonal antibody. Plot shows effects of indicated mutations on antibody (x-axis) and ACE2 (y-axis) binding26. Mutations are classed according to whether they are accessible by single mutation from the Wuhan-1 strain (single-nt accessible) and those that require multi-nucleotide mutations. b, Neutralization of SARS-CoV-2 variants by S2E12 (spike-pseudotyped VSV on Vero E6 cells), relative to Wuhan-1 D614G, as in Fig. 2d. c, S2E12 breadth of neutralization (spike-pseudotyped VSV on 293T-ACE2 cells). Points represent mean of biological duplicates. d, Replicative fitness of S2E12 escape mutations identified in a on Vero E6 cells. Data are mean ± s.e.m. from triplicate experiments. e, f, Structures of S2E12 Fab (e) and S2D106 Fab (f) bound to SARS-CoV-2 RBD. RBD sites coloured by escape (colour bar, centre). The E484 side chain is included for visualization purposes only but was not included in the final S2D106-bound structure owing to weak density. Max, maximum. g, h, Integrative features of the structural footprints (5 Å cut-off) of S2E12 (g) and S2D106 (h). Sites are ordered by the frequency of observed mutants among SARS-CoV-2 sequences on GISAID. Heat maps as in Fig. 2b. Logo plots as in Fig. 1c, but showing only amino acid mutations accessible via single-nucleotide mutation from Wuhan-1 strain for comparison with a. Bar plots illustrate frequency of SARS-CoV-2 mutants and their validated effects on antibody neutralization (spike-pseudotyped VSV on Vero E6 cells). Red indicates greater than tenfold increase in IC50 due to mutation. ND, not determined.
Many RBM antibodies such as S2X58 and S2D106 select mutations present in SARS-CoV-2 variants of concern (for example, L452R and E484K)27,28,29. By contrast, S2E12 selects viral mutants at sites that do not exhibit substantial variation among circulating SARS-CoV-2, and S2E12 correspondingly neutralizes a diverse panel of SARS-CoV-2 variants17 (Fig. 3b). S2E12 is also unique in its breadth among RBM antibodies (Fig. 1d), neutralizing VSV pseudotyped with each of the four SARS-CoV-2 clade sarbecovirus spikes (Fig. 3c, Extended Data Fig. 4j). As with S2H97, somatic mutations in S2E12 enhanced affinity across sarbecoviruses, though the increase in affinity was more modest than for S2H97 (Extended Data Fig. 4k, l).
Conservation of the S2E12 epitope among SARS-CoV-2 variants could reflect the relative rarity of S2E12-like antibodies in polyclonal sera leading to little antigenic pressure at these sites (Fig. 2g), together with functional constraint in the S2E12 epitope (escapability being the lowest for S2E12 and S2H97 among the 12 antibodies evaluated). Indeed, the strong antibody-escape mutations that emerged in S2E12 viral escape selections decrease ACE2 binding affinity26 (Fig. 3a) and reduce replicative fitness in a bulk competition experiment between spike-expressing VSV variants passaged in the absence of antibody (Fig. 3d).
To understand the structural basis for the unique breadth and robustness of S2E12 to escape, we compared its structure to that of S2D106 Fab (cryoEM, 4.0 Å resolution local refinement) bound to SARS-CoV-2 RBD (Fig. 3e, f, Extended Data Fig. 5g, h, m–p, Extended Data Tables 2, 3). We also integrated evolutionary, functional, and structural details for the sites in each antibody’s structural footprint (Fig. 3g, h). S2E12 and S2D106 bind the receptor-binding ridge, with 8 residues shared between their footprints. S2E12 binding is oriented towards extensive packing of the ACE2-contact residue F486RBD within a cavity lined by aromatic residues at the antibody light–heavy-chain interface (Fig. 3e, Extended Data Fig. 5g), as was seen with the homologous antibody COV2-219636. Sites within the S2E12 footprint that exhibit less functional constraint (for example, E484 and S477) are located at the periphery of the interface, explaining the robustness of S2E12 towards SARS-CoV-2 variants (Fig. 3b, g). This structural interface also explains the breadth of S2E12 towards RaTG13 and GX-pangolin-CoV (Fig. 1d), as the F486L mutation present in these sarbecoviruses retains the central hydrophobic packing.
In contrast to S2E12, S2D106 binding is centred on residue E484RBD which may form a salt bridge with R96LC, in addition to nonpolar contacts between F490RBD and residues in the heavy chain CDR2 (Fig. 3f, Extended Data Fig. 5h). Although the long heavy chain CDR3 packs intimately across the surface of the RBD, there are no crucial CDRH3–RBD contacts that are sensitive to mutation. S2D106 escape is therefore highly focused on E484 and F490, which are functionally tolerant and exhibit variation among SARS-CoV-2 sequences (Fig. 3h). This comparison between S2E12 and S2D106 highlights how small differences in the RBD–antibody interface affect the breadth and robustness of each antibody to viral escape.
The landscape of RBD epitopes
Last, we examined how escapability, breadth, and neutralization potency relate to one another and to the RBD epitope. We used our binding-escape maps (Fig. 1b, c), together with comparable maps published for other RBD antibodies3,20,21,36,37, to project antibodies into a two-dimensional space on the basis of similarities in sites of binding-escape mutations (Fig. 4a).

a, Multidimensional scaling projection of similarities in antibody binding-escape maps from the present (red) and previous (grey) studies. Pie charts illustrate the RBD subdomains where mutations confer escape (key, bottom left: RBM, residues 437–508; ACE2 contact, 4 Å cutoff). Structural projections of escape arrayed around the perimeter (colour bar, bottom right), with grey outlines tracing structural footprints. b–d, Projected epitope space from a annotated by antibody properties: SARS-CoV-2-neutralizing potency (b), sarbecovirus-binding breadth (c) and escape by natural SARS-CoV-2 mutants (d). For each property, antibodies are coloured such that purple reflects the most desirable antibody (colour bar, right): most potent neutralization (log10 scale), highest breadth, and lowest natural frequency of escape mutants (log10 scale). e, Relationship between SARS-CoV-2 neutralization potency and sarbecovirus breadth for antibodies described in this work and in a parallel study37 (S2X259). f, Relationship between functional epitope size and SARS-CoV-2 RBD binding affinity. g, Relationship between natural SARS-CoV-2 escape mutant frequency (Extended Data Fig. 3c) and sarbecovirus breadth.
We annotated our projection of epitope space using antibody properties such as in vitro neutralization potency, breadth and escapability (Fig. 4b–d, Extended Data Fig. 7c, d). The most potently neutralizing antibodies (for example, S2E12 and S2D106) bind epitopes in the RBM, whereas antibodies targeting the core RBD are less potently neutralizing (Fig. 4b). Of note, RBD antibodies can protect in vivo through other mechanisms beyond neutralization12,13,22. Antibodies with broad sarbecovirus binding target the core RBD (Fig. 4c). Our panel therefore extends previous observations4,5,32,38 to highlight a general trade-off between sarbecovirus breadth and potency of SARS-CoV-2 neutralization (Fig. 4e). Nonetheless, some cross-reactive antibodies exhibit intermediate in vitro neutralization potency (for example, S309 and S2X25937), and the highly potent RBM-directed antibody S2E12 exhibits modest breadth, highlighting the existence of antibodies that balance neutralization potency and breadth.
The size of an antibody’s functional epitope (Fig. 1b, c) is not strongly influenced by the structural location of the epitope (Extended Data Fig. 7c)—instead, narrower functional epitopes are associated with higher Fab–RBD binding affinity (Fig. 4f). However, an antibody’s escapability, which integrates how escape mutations affect RBD folding and ACE2 affinity, is influenced by variation in these functional constraints across the RBD structure. For example, antibodies that cluster with S2E12 exhibit lower escapability (Extended Data Fig. 7c) and frequency of natural SARS-CoV-2 escape mutants (Fig. 4d). As highlighted by S2E12 and S2H97 above, antibodies with even a modest degree of sarbecovirus breadth show a greatly reduced frequency of escape mutations among circulating SARS-CoV-2 variants (Fig. 4g).
Principles for antibody and vaccine development
Ongoing SARS-CoV-2 evolution19,27,28,29, long-term antigenic evolution of other human coronaviruses39,40 and the spillover potential of diverse sarbecovirus lineages6,7 indicate the importance of developing antibodies and vaccines that are robust to viral evolution. In this work, we identify antibody and epitope features that can guide this process. Although in vitro neutralization potency is often prioritized for lead selection, our results suggest that this will bias antibodies towards RBM epitopes, many of which are poorly conserved in the short-term evolution of SARS-CoV-219 and the long-term evolution of sarbecoviruses7. Our results suggest that additional prioritization of high-affinity binding and at least a moderate degree of sarbecovirus breadth will yield antibodies with improved resistance to viral escape4,5.
A long-term goal is to develop antibodies and vaccines that cross-react with distant sarbecovirus lineages capable of zoonotic spillover. We have identified a cryptic epitope capable of eliciting pan-sarbecovirus immunity, represented by S2H97. Though S2H97-like antibodies are rare in polyclonal sera, the protective capacity and exceptional breadth of S2H97 indicates that pan-sarbecovirus vaccines could seek to improve responses to this epitope by unmasking this and other cryptic broadly neutralizing epitopes5,37,41. Broader cross-reactivity among betacoronavirus lineages including MERS and OC43 has been reported for antibodies that bind the spike S2 domain32,38,42. Though S2H97 breadth does not extend beyond sarbecoviruses, its discovery expands our view of what can be achieved via a potent RBD-directed antibody response.
The global emergence of variants of concern (VOC) has been an important feature of the pandemic27,28,29. Mutations in VOC occur in immunodominant RBM epitopes (for example, residues E484, K417 and L452) and influence binding by polyclonal serum and some therapeutic antibodies17,18,19,20,21. We cannot predict which mutations will next rise to prominence as SARS-CoV-2 continues to evolve, but it seems likely that they will include additional RBM mutations that affect recognition by infection- and vaccine-elicited antibodies1,2,15,16,19. Therefore, antibody discovery efforts focused on breadth4,5, aided by high-resolution differentiation among antibody epitopes as generated here, can inform the development of antibody and vaccine countermeasures with greater robustness to immune escape in the current SARS-CoV-2 pandemic and utility for potential future sarbecovirus spillovers.
Methods
Mammalian cell lines
Cell lines were received from ATCC (Vero E6, Vero, BHK-21, CHO-K1, HEK293T/17), Takara (Lenti-X 293T) and Thermo Fisher Scientific (ExpiCHO-S, Expi293F and Freestyle 293-F). MA104 cells were a gift from H. B. Greenberg (Stanford School of Medicine). The 293T-ACE2 cells are described in references31,43. Vero and MA104 cell lines tested negative for mycoplasma contamination. Other cell lines were not tested. No authentication was performed beyond manufacturer standards.
Isolation of peripheral blood mononuclear cells, plasma and sera
Samples from three individuals who had recovered from SARS-CoV-2, designated as donors S2H (age 36, male), S2D (age 70, male) and S2X (age 52, male) were obtained under study protocols approved by the local Institutional Review Board (Canton Ticino Ethics Committee, Switzerland). All donors provided written informed consent for the use of blood and blood components (such as peripheral blood mononuclear cells (PBMCs), sera or plasma). Blood drawn from donor S2X was obtained at day 48 (S2X16, S2X35 and S2X58 antibodies) and 75 (S2X227) after symptoms onset. Blood from donor S2H was obtained at day 17 (S2H13 and S2H14), day 45 (S2H58) and day 81 (S2H97) after symptoms onset. Blood from donor S2D was obtained at day 98 (S2D106) after symptoms onset.
PBMCs were isolated from blood draw performed using tubes pre-filled with heparin, followed by Ficoll density gradient centrifugation. PBMCs were either used fresh for SARS-CoV-2 Spike protein-specific memory B cell sorting or stored in liquid nitrogen for later use. Sera were obtained from blood collected using tubes containing clot activator, followed by centrifugation and storage at −80 °C.
Sera for blockade of binding serological assays were obtained from 3 cohorts of SARS-CoV-2 convalescent (average age 52, range 25–78, 55% male) or vaccinated (average age 49, range 28–69, 65% male) individuals under study protocols approved by the local Institutional Review Boards (Canton Ticino Ethics Committee, Switzerland, the Ethical Committee of Luigi Sacco Hospital, Milan, Italy, and WCG North America, Princeton, NJ, USA). All donors provided written informed consent for the use of blood and blood components (such as PBMCs, sera or plasma) and were recruited at hospitals or as outpatients.
B-cell isolation and recombinant monoclonal antibody production
The discovery and initial characterization of six of the antibodies in our panel has been previously reported (S309 and S304, refs. 4,15; S2X35, S2H13 and S2H14, ref. 15; and S2E12, ref. 8), and the other six (S2H97, S2X16, S2H58, S2D106, S2X58 and S2X227) are described for the first time in this article. Starting from freshly isolated PBMCs or upon cell thawing, B cells were enriched by staining with CD19 PE-Cy7 (BD Bioscience 557835, 1:50) and incubation with anti-PE MicroBeads (Miltenyi Biotec 130-048-801, 1:100), followed by positive selection using LS columns (Miltenyi Biotec). Enriched B cells were stained with anti-IgM (BioLegend 314508, 1:20), anti-IgD (BD Bioscience 555779, 1:40), anti-CD14 (BD Bioscience 562691, 1:50) and anti-IgA (Southern Biotech 2050-09, 1:400), all PE labelled, and prefusion SARS-CoV-2 S with a biotinylated Avi-tag (in house produced) conjugated to Streptavidin–Alexa-Fluor 647 (Life Technologies S21374, 1:40). SARS-CoV-2 S-specific IgG+ memory B cells were sorted by flow cytometry via gating for PE negative and Alexa-Fluor 647 positive cells. Cells were cultured for the screening of positive supernatants. Antibody VH and VL sequences were obtained by PCR with reverse transcription (RT–PCR) and monoclonal antibodies were expressed as recombinant human Fab fragment or as IgG1 (G1m3 allotype) carrying the half-life-extending M428L/N434S (LS) mutation in the Fc region. ExpiCHO-S cells (Thermo Fisher Scientific) were transiently transfected with heavy and light chain expression vectors as previously described4. Affinity purification was performed on Äkta Xpress FPLC (Cytiva) operated by Unicorn software version 5.11 (Build 407) using HiTrap Protein A columns (Cytiva) for full length human monoclonal antibodies and CaptureSelect CH1-XL MiniChrom columns (Thermo Fisher Scientific) for Fab fragments, using PBS as mobile phase. Buffer exchange to the appropriate formulation buffer was performed with a HiTrap Fast desalting column (Cytiva). The final products were sterilized by filtration through 0.22-μm filters and stored at 4 °C.
Using the Database IMGT (http://www.imgt.org), the Vh and Vl germline gene family and the number of somatic mutations were determined by analysing the homology of the Vh and Vl sequences to known human V, D and J genes. Germline-reverted sequences of the Vh and Vl were constructed using IMGT/V-QUEST. The S2E12 and S2H97 germline-reverted antibodies (G1m17 allotype) were produced by ATUM. Germline-reverted Fab fragments were generated by digestion of the corresponding IgGs.
Epitope classes shown in Figs. 1a, 2g are defined as in Piccoli et al. 15. In brief, the classification of these epitope classes results from Octet binning experiments using structurally characterized antibodies, structural insights to define the recognition of open-only RBD and ability of antibodies to interfere with RBD binding to ACE2. In particular, site Ia is accessible only in the open state of RBD and largely overlaps the ACE2 footprint; site Ib is accessible in both open and closed RBD states and overlaps in part the ACE2 footprint; site IIa is in the core RBD (accessible only in the open RBD state) and antibodies binding to this site interfere with binding to ACE2, site IIc is also in the core RBD but targeted by antibodies that do not interfere with binding to ACE2; site IV is fully accessible on both open and closed RBDs and is defined by the footprint of the S309 antibody.
Neutralization of authentic SARS-CoV-2 by entry-inhibition assay
Neutralization was determined using SARS-CoV-2-Nluc, an infectious clone of SARS-CoV-2 (based on strain 2019-nCoV/USA_WA1/2020) which encodes nanoluciferase in place of the viral ORF7 and demonstrates comparable growth kinetics to wild-type virus44. Vero E6 cells (ATCC, CRL-1586) were seeded into black-walled, clear-bottom 96-well plates at 2 × 104 cells per well and cultured overnight at 37 °C. The next day, 9-point fourfold serial dilutions of monoclonal antibodies were prepared in infection media (DMEM + 10% FBS). SARS-CoV-2-Nluc was diluted in infection media at a final multiplicity of infection (MOI) of 0.01 plaque-forming units (PFU) per cell, added to the monoclonal antibody dilutions and incubated for 30 min at 37 °C. Medium was removed from the Vero E6 cells and monoclonal antibody–virus complexes were added and incubated at 37 °C for 24 h. Media was removed from the cells, Nano-Glo luciferase substrate (Promega) was added according to the manufacturer’s recommendations, incubated for 10 min at room temperature and the luciferase signal was quantified on a Victor Nivo plate reader (Perkin Elmer).
SARS-CoV-2 spike pseudotyped VSV generation and neutralization assay
Replication-defective VSV pseudoviruses45 expressing SARS-CoV-2 spike protein were generated as previously described46 with some modifications. Plasmids encoding SARS-CoV-2 spike single-mutant variants were generated by site-directed mutagenesis of the wild-type plasmid, pcDNA3.1(+)-spike-D1947, and plasmids encoding multiply mutated SARS-CoV-2 variants of concern were generated using a multistep overlap extension PCR protocol23,48, in which sequential, overlapping fragments were designed to introduce all mutations, which were PCR assembled and cloned into the pcDNA3.1 vector using the Takara In-fusion HD cloning kit following manufacturer’s instructions.
Lenti-X 293T (Takara, 632180) cells were seeded in 10-cm dishes at a density of 1 × 105 cells per cm2 and the following day transfected with 5 μg of spike expression plasmid with TransIT-Lenti (Mirus, 6600) according to the manufacturer’s instructions. For the neutralization assays with variants of concern (Figs. 2d, 3b), Lenti-X 293T cells were seeded in 10-cm dishes at a density of 5 × 106 cells per cm2, and transfected the following day with 10 μg of spike expression plasmid. One day after transfection, cells were infected with VSV (G*ΔG-luciferase) (Kerafast, EH1020-p.m.) for 1 h, rinsed three times with PBS, then incubated for an additional 24 h in complete medium at 37 °C. The cell supernatant was clarified by centrifugation, filtered (0.45 μm), aliquoted, and frozen at −80 °C.
For VSV pseudovirus neutralization assays, Vero E6 cells (ATCC, CRL-1586) were grown in DMEM supplemented with 10% FBS and seeded into clear bottom white 96 well plates (Costar, 3903) at a density of 2 × 104 cells per well. The next day, monoclonal antibodies were serially diluted in pre-warmed complete media, mixed at a 1:1 ratio with pseudovirus and incubated for 1 h at 37 °C in round bottom polypropylene plates. Media from cells was aspirated and 50 μl of virus–monoclonal antibody complexes were added to cells and then incubated for 1 h at 37 °C. An additional 100 μl of prewarmed complete media was then added on top of complexes and cells incubated for an additional 16–24 h. Conditions were tested in duplicate wells on each plate and at least six wells per plate contained uninfected, untreated cells (mock) and infected, untreated cells (‘no mAb control’).
Medium containing virus–monoclonal antibody complexes was then aspirated from cells and 100 μl of a 1:4 dilution of Bio-glo (Promega, G7940) in PBS was added to cells. For neutralization assays with variants of concern, 50 μl of a 1:2 dilution of SteadyLite Plus (Perkin Elmer) in PBS with Ca2+ and Mg2+ was added to cells in place of Bio-glo. Plates were incubated for 10 min at room temperature and then were analysed on an Envision plate reader (PerkinElmer), or for variants of concern assays, a Synergy H1 Hybrid Multi-Mode reader (Biotek).
Relative light unit (RLU) measurements for infected wells were subtracted by the average of RLU values for the mock wells (background subtraction) and then normalized to the average of background subtracted ‘no mAb control’ RLU values within each plate. Per cent neutralization was calculated by subtracting from 1 the normalized monoclonal antibody infection condition. Data were analysed and visualized with Prism (version 8.4.3). IC50 values were calculated from the interpolated value from the log(inhibitor) versus response–variable slope (four parameters) nonlinear regression with an upper constraint of <100. Neutralization experiments with wild-type SARS-CoV-2 S and single-mutant variants were conducted on three independent days, that is, biological replicates, where each biological replicate contains a technical duplicate. IC50 values across biological replicates are presented as geometric mean. The loss or gain of neutralization potency across spike variants was calculated by dividing the variant IC50 by the parental IC50 within each biological replicate. Neutralization experiments with SARS-CoV-2 S variants of concern were conducted in biological duplicates, with IC50 values normalized by the corresponding wild-type measurement, and presented as arithmetic mean of the duplicate experiments.
SARS-CoV-2 spike pseudotyped VSV neutralization on 293T-ACE2 cells
To investigate the effect of ACE2 expression on S2H97 neutralization, Vero E6 cells were seeded at 20,000 cells per well in black clear-bottom 96-well plates. 293T-ACE2 cells31 were seeded at 35,000 cells per well in black clear-bottom 96-well plates that had been pre-coated with poly-d-lysine (Gibco). The next day, SARS-CoV-2 spike-pseudotyped VSV neutralizations with S2E12, S309 and S2H97 were performed as described above. Neutralizations were performed in triplicate wells.
Sarbecovirus spike pseudotyped VSV neutralization by S2H97
Mammalian expression constructs (pcDNA3.1(+) or pTwist-CMV) encoding the spike proteins from various sarbecoviruses with a C-terminal deletion of 19 amino acids (D19) were synthesized for SARS-CoV-2 (GenBank: QOU99296.1), SARS-CoV-1 Urbani (GenBank: AAP13441.1), hCoV-19/pangolin/Guangdong/1/2019 (GD-pangolin-CoV, GenBank: QLR06867.1), pangolin coronavirus Guanxi-2017 (GX-pangolin-CoV, GenBank: QIA48623.1), and bat sarbecovirus WIV1 (WIV1, GenBank: AGZ48828.1). Lenti-X 293T cells (Takara, 632180) were seeded in 15-cm dishes such that the cells would reach 80% confluency after culturing overnight. The following day, cells were transfected using TransIT-Lenti (Mirus, 6600) according to the manufacturer’s instructions. One day after transfection, cells were infected with VSV (G*ΔG-luciferase) (Kerafast, EH1020-p.m.). The supernatant containing sarbecovirus pseudotyped VSV was collected 2 days after transfection, centrifuged at 1,000g for 5 min, aliquoted and frozen at −80 °C.
For neutralization assays, cells supporting robust pseudovirus infection were seeded into clear bottom white-walled 96-well plates at 20,000 cells per well in 100 μl culture media. Vero E6 cells were used for VSV-SARS-CoV-2, VSV-SARS-CoV-1, and VSV-GD-pangolin-CoV. BHK-21 cells (ATCC, CCL-10) stably expressing ACE2 were used for VSV-GX-pangolin-CoV and VSV-WIV1. After culturing cells overnight, 1:3 serial dilutions of antibody were prepared in DMEM in triplicate. Pseudovirus was diluted in DMEM and added to each antibody dilution such that the final dilution of pseudovirus was 1:20. Pseudovirus–antibody mixtures were incubated for 1 h at 37 °C. Medium was removed from the cells and 50 μl of pseudovirus:antibody mixtures were added. One hour after infection, 50 μl of culture medium was added to wells containing pseudovirus:antibody mixtures and incubated overnight at 37 °C. Medium was then removed and 100 μl of 1:1 diluted DPBS:Bio-Glo (Promega, G7940) luciferase substrate was added to each well. The plate was shaken at 300 rpm at room temperature for 10 min after which RLU reading were taken on an EnSight (Perkin Elmer) microplate reader. Per cent neutralization was determined by first subtracting the mean background (cells with luciferase substrate alone) RLU values of 6 wells per plate for all data points. Per cent neutralization for each antibody concentration was calculated relative to no-antibody control wells for each plate. Per cent neutralization data were analysed and graphed using Prism (GraphPad, v9.0.1). Absolute IC50 values were calculated by fitting a curve using a nonlinear regression model (variable slope, 4 parameters) and values were interpolated from the curve at y = 50%. The geometric mean from at least two independent experiments was calculated using Excel (Microsoft, version 16.45).
Sarbecovirus spike pseudotyped VSV neutralization by S2E12
Spikes from SARS-CoV-2 (CAD0240757.1), RaTG13 (QHR63300.2), GD-pangolin (QLR06867.1), GX-pangolin (QIA48623.1), SARS-CoV-1 Tor2 (YP009825051), WIV1 (AGZ48831.1) and WIV16 (ALK02457.1) were used to pseudotype VSV. To produce pseudotyped viruses, HEK293T/17 (ATCC, CRL-11268) seeded in 10-cm dishes in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin were transfected with plasmids using lipofectamine 2000 (Life Technologies) following manufacturer’s instructions. One day post-transfection, cells were infected with VSV (G*ΔG-luciferase) for 2 h and washed 4 times with DMEM, before adding medium supplemented with anti-VSV-G antibody (I1-mouse hybridoma supernatant at 1:50 dilution, from CRL-2700, ATCC). Pseudotyped particles were collected 18 h after inoculation, clarified by centrifugation at 2,000g for 5 min, concentrated 10× with a 30 kDa cut-off membrane filter, and stored at −80 °C. For S2E12 neutralization experiments, 293T cells stably expressing ACE2 (BEI #NR-52511)43 in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin were seeded at 40,000 cells per well in clear-bottom white-walled 96-well plates and cultured overnight at 37 °C. Twelve threefold serial dilutions of S2E12 antibody were prepared in DMEM, and antibody dilutions were mixed 1:1 with pseudotyped VSV in the presence of 1:50 diluted anti-VSV-G antibody. After 45 min incubation at 37 °C, 40 μl of antibody–virus mixture was added to cells, and 40 μl DMEM was added 2 h post-infection. After 17–20 h, 50 μl One-Glo-EX substrate (Promega) was added to the cells. Cells were incubated in the dark for 5–10 min before luminescence reading on a Varioskan LUX plate reader (Thermo Fisher Scientific). Relative luciferase unit values were converted to percentage of neutralization and plotted with a nonlinear regression curve fit in GraphPad Prism. Measurements were performed in duplicate with two independent productions of pseudotyped virus.
Recombinant protein production
SARS-CoV-2 RBD wild-type protein for SPR binding assays (with N-terminal signal peptide and C-terminal thrombin cleavage site-TwinStrep-8×His-tag) was expressed in Expi293F (Thermo Fisher Scientific) cells at 37 °C and 8% CO2. Transfections were performed using the ExpiFectamine 293 Transfection Kit (Thermo Fisher Scientific). Cell culture supernatants were collected three days after transfection and supplemented with 10× PBS to a final concentration of 2.5× PBS (342.5 mM NaCl, 6.75 mM KCl and 29.75 mM phosphates). SARS-CoV-2 RBDs were purified using 1 or 5 ml HisTALON Superflow cartridges (Takara Bio) and subsequently buffer exchanged into 1× HBS-N buffer (Cytiva) or PBS using a Zeba Spin Desalting (Thermo Fisher Scientific) or HiPrep 26/10 (Cytiva) desalting column.
SARS-CoV-2 RBD wild-type for crystallization (with N-terminal signal peptide, ETGT and C-terminal 8×His-tag) was expressed similarly as described above in the presence of 10 μM kifunensine. Cell culture supernatant was collected four days after transfection and supplemented with 10× PBS to a final concentration of 2.5× PBS. Protein was purified using a 5 ml HisTALON Superflow cartridge followed by size-exclusion chromatography on a Superdex 200 Increase 10/300 GL column (Cytiva) equilibrated in 20 mM Tris-HCl pH 7.5, 150 mM NaCl. For crystallization of the RBD-S2X259-S2H97 and RBD-S2E12-S304-S309 Fab complexes, RBD was deglycosylated by overnight incubation with EndoH glycosidase at 4 °C.
RBDs from other sarbecoviruses for SPR (with N-terminal signal peptide and C-terminal thrombin cleavage site-TwinStrep-8×His-tag) were expressed in Expi293F cells at 37 °C and 8% CO2. Cells were transfected using PEI MAX (Polysciences) at a DNA:PEI ratio of 1:3.75. Transfected cells were supplemented three days after transfection with 3 g l−1 glucose (Bioconcept) and 5 g l−1 soy hydrolysate (Sigma-Aldrich). Cell culture supernatant (423 ml) was collected 7 days after transfection and supplemented with 47 ml 10× binding buffer (1 M Tris-HCl, 1.5 M NaCl, 20 mM EDTA, pH 8.0) and 25 ml BioLock (IBA) and incubated on ice for 30 min. Proteins were purified using a 5-ml Strep-Tactin XT Superflow high-capacity cartridge (IBA) followed by buffer exchange to PBS using HiPrep 26/10 desalting columns (Cytiva).
Prefusion-stabilized SARS-CoV-2 spike proteins for SPR (residues 14-1211, either D614 or D614G), containing the 2P and Furin cleavage site mutations49 with a mu-phosphatase signal peptide and a C-terminal Avi-8×His-C-tag or C-terminal 8xHis-Avi-C-tag were expressed in Freestyle 293-F cells (Thermo Fisher Scientific, R79007) at 37 °C and 8% CO2. Transfections were performed using 293fectin as a transfection reagent. Cell culture supernatant was collected after three days and purified over a 5 ml C-tag affinity matrix. Elution fractions were concentrated and injected on a Superose 6 Increase 10/300 GL column (Cytiva) with 50 mM Tris-HCl pH 8.0 and 200 mM NaCl as running buffer.
SARS-CoV-2 HexaPro spike protein for cryoEM analysis was produced in Freestyle 293-F cells grown in suspension using FreeStyle 293 expression medium (Life Technologies) at 37 °C in a humidified 8% CO2 incubator rotating at 130 rpm. The cultures were transfected using PEI (9 μg ml−1) with cells grown to a density of 2.5 million cells per ml and cultivated for three days. The supernatants were collected and cells resuspended for another three days, yielding two collections from each such culture. Spike proteins were purified from clarified supernatants using a 5 ml cobalt affinity column (Cytiva, HiTrap TALON crude), concentrated and flash frozen in a buffer containing 20 mM Tris pH 8.0 and 150 mM NaCl before analysis.
SARS-CoV-2 S native-like ectodomain trimer for refolding assays was engineered with a mu-phosphatase signal peptide beginning at 14Q, a mutated S1/S2 cleavage site (SGAR), and a TEV cleavage, fold-on trimerization motif, and 8x His tag appended to the C terminus (K1211). Native-like spike was expressed and purified as described for SARS-CoV-2 HexaPro spike above.
Recombinant hACE2 for SPR (residues 19–615 from Uniprot Q9BYF1 with a C-terminal AviTag-10×His-GGG-tag, and N-terminal signal peptide) was produced by ATUM. Protein was purified via Ni Sepharose resin followed by isolation of the monomeric hACE2 by size exclusion chromatography using a Superdex 200 Increase 10/300 GL column (Cytiva) pre-equilibrated with PBS.
SPR binding assays
SPR binding measurements were performed using a Biacore T200 instrument with CM5 sensor chip covalently immobilized with StrepTactin XT to capture recombinant RBD proteins (data in Fig. 1a, Extended Data Figs. 1c, 4f, i, l). Running buffer was Cytiva HBS-EP+ (pH 7.4). All measurements were performed at 25 °C. Fab (or hACE2) analyte concentrations were 11, 33, 100 and 300 nM, run as single-cycle kinetics. Double reference-subtracted data were fit to a 1:1 binding model using Biacore T200 Evaluation (version 3.1) or Biacore Insight Evaluation (version 2.0.15) software. KD values above 1 μM were determined from fits where the maximum SPR signal at saturation (Rmax) was set as a constant determined from results for higher affinity analytes binding to the same RBD at the same surface density. Data where averages are not given are representative of duplicate or triplicate measurements (except measurements with germline Fabs, which were singleton measurements).
To corroborate the SARS-CoV-2 RBD binding measurements, experiments were also performed in two additional formats, both with monovalent analytes (data in Extended Data Table 1): (1) Fab binding to SARS-CoV-2 spike ectodomain was measured using CM5 sensor chips immobilized with anti-AviTag pAb (Genscript, A00674-40) for capturing S, other experiment parameters same as above, and (2) RBD binding to IgG was measured using CM5 sensor chips immobilized with anti-human Fc pAb (Southern Biotech, 2014-01) for capturing IgG, with RBD analyte concentrations of 3.1, 12.5, and 50 nM, other experiment parameters same as above. Fit results yield an apparent KD for the spike-binding experiments because the kinetics also reflect spike conformational dynamics. Spike ectodomain was D614G with C-terminal 8xHis-Avi-C-tag for all measurements except S2X58 binding was performed with D614 spike with C-terminal Avi-8×His-C-tag. For the comparison of mature and germline-reverted antibody binding to RaTG13, the data reported are from experiment format (2) with IgG as ligand. These data (and others indicated in Extended Data Table 1 as 'biphasic kinetics') were fit to a heterogeneous ligand model, due to an artefactual kinetic phase with very slow dissociation that often arises when RBD is an analyte; the lower affinity of the two KD values reported by the fit is given as the KD (the two KD values are separated by at least one order of magnitude).
Deep mutational scanning mutant escape profiling
We used a previously described deep mutational scanning approach3 to comprehensively identify RBD mutations that escape binding by each antibody. This approach leverages duplicate RBD mutant libraries26, which contain virtually all of the 3,819 possible amino acid mutations in the background of the Wuhan-Hu-1 RBD sequence. Library variants were previously linked to short identifier barcode sequences and sorted to purge the library of variants that strongly decrease ACE2 binding affinity or expression of folded RBD3.
We first used an isogenic yeast strain expressing the unmutated SARS-CoV-2 RBD and flow cytometry to identify the EC90 of each antibody’s binding to yeast-displayed SARS-CoV-2 RBD. We then performed library selections as previously described3,20, labelling libraries with the EC90 concentration of antibody to standardize escape mutation sensitivity across selections. In brief, libraries of yeast were induced for surface expression, washed, and labelled with the primary antibody for 1 h at room temperature. Cells were washed, and secondarily labelled with 1:200 PE-conjugated goat anti-human-IgG antibody (Jackson ImmunoResearch 109-115-098) to label for bound antibody, and 1:100 FITC-conjugated chicken anti-Myc-tag (Immunology Consultants Lab, CYMC-45F) to label for RBD surface expression. We prepared controls for setting FACS selection gates by labelling yeast expressing the unmutated SARS-CoV-2 RBD with the same antibody concentration as library selections (1×), 100× reduced antibody concentration to illustrate the effect of mutations with 100×-reduced affinity, and 0 ng ml−1 antibody to illustrate complete loss of antibody binding. Representative selection gates are shown in Extended Data Fig. 2b. Gates were set and sorting performed with FACSDiva software (version 6.1.3). We sorted approximately 7.5 × 106 RBD+ cells per library on a BD FACSAria II, collecting yeast cells from the antibody-escape sort bin (fractions of library falling into antibody escape bin given in Extended Data Fig. 2c). Sorted cells were recovered overnight, plasmids were extracted from the pre-sort and antibody-escape populations, and variant-identifier barcode sequences were PCR amplified and sequenced on an Illumina HiSeq 25003,26.
As previously described3,20, sequencing counts pre- and post-selection were used to estimate the ‘escape fraction’ for each library variant, which reflects the fraction of yeast expressing a variant that fall into the antibody-escape FACS bin. In brief, we used the dms_variants package (https://jbloomlab.github.io/dms_variants/, version 0.8.2) to process Illumina sequences into variant counts pre- and post-selection using the barcode/RBD variant lookup table from Starr et al. 26. We then computed per-variant escape fraction of variant v (Ev) as: \({E}_{v}=F\times ({n}_{{v}_{{\rm{p}}{\rm{o}}{\rm{s}}{\rm{t}}}}/{N}_{{\rm{p}}{\rm{o}}{\rm{s}}{\rm{t}}})/({n}_{{v}_{{\rm{p}}{\rm{r}}{\rm{e}}}}/{N}_{{\rm{p}}{\rm{r}}{\rm{e}}})\), where F is the total fraction of the library that escapes antibody binding (Extended Data Fig. 2c), \({n}_{{v}_{{\rm{p}}{\rm{o}}{\rm{s}}{\rm{t}}}}\) and \({n}_{{v}_{{\rm{p}}{\rm{r}}{\rm{e}}}}\) are the sequencing counts of variant v in the RBD library after and before FACS selection, respectively (with a pseudocount of 0.5 added to all counts), and Npost and Npre are the total counts of all variants after and before FACS selection, respectively. We then applied computational filters to remove variants with low pre-sort sequencing counts or highly deleterious mutations that might cause artefactual antibody escape due to global unfolding or loss of expression of RBD on the cell surface. Specifically, we filtered out variants whose pre-selection sequencing counts were lower than the 99th percentile counts of variants containing premature stop codons, which were largely purged by the prior sorts for RBD expressing and ACE2-binding RBD variants. We also removed variants with ACE2 binding scores below −2.35 or RBD expression scores below −1, and variants containing individual mutations with effects below these thresholds, using the variant- and mutation-level deep mutational scanning measurements of Starr et al. 26. We also filtered out rare mutations with low coverage in the libraries, retaining mutations that were sampled on at least one single-mutant barcoded variant or at least two multiply mutated variants in each replicate. Last, to decompose single-mutation escape fractions for each antibody, we implemented global epistasis models50 using the dms_variants package to estimate the effect of each individual amino acid mutation, exactly as described in ref. 20.
Antibody escape selections were conducted in full duplicate using independently generated and assayed SARS-CoV-2 mutant libraries (see correlations in Extended Data Fig. 2e, f). The reported escape fractions throughout the paper are the average across the two replicates, and these final per-mutation escape fractions are provided on GitHub (https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_Vir_mAbs/blob/main/results/supp_data/vir_antibodies_raw_data.csv). Interactive visualizations of antibody escape maps (https://jbloomlab.github.io/SARS-CoV-2-RBD_MAP_Vir_mAbs) were created using dms-view51.
Sarbecovirus library binding assays
A curated set of all unique sarbecovirus RBD amino acid sequences was gathered, including the sarbecovirus RBD sequence set reported by Letko et al. 7, along with additional unique RBD sequences among SARS-CoV-1 epidemic strains reported by Song et al. 52, BtKY7253 and new sarbecovirus sequences RmYN0254, GD-pangolin-CoV (consensus RBD reported in figure 3a of Lam et al. 55), and GX-pangolin-CoV55 (P2V, ambiguous nucleotide within codon 515 (SARS-CoV-2 spike numbering) resolved to retain F515, which is conserved in all other sarbecoviruses). A list of all RBDs and sequence accession numbers is available on GitHub (https://github.com/jbloomlab/SARSr-CoV_RBD_MAP/blob/main/data/RBD_accessions.csv).
To define clades of sarbecovirus RBDs, an alignment of amino acid RBD sequences was generated using mafft56 with gap opening penalty 4.5 (alignment available on GitHub: https://github.com/jbloomlab/SARSr-CoV_RBD_MAP/blob/main/data/RBD_aa_aligned.fasta). The corresponding nucleotide sequence alignment was generated from the amino acid alignment using PAL2NAL57. The gene sequence phylogeny was inferred using RAxML version 8.2.1258, with the GTRGAMMA substitution model and a partition model with separate parameters for first, second and third codon positions. The Hibecovirus RBD sequence Hp-Zhejiang2013 (GenBank: KF636752) was used as an outgroup for rooting of the sarbecovirus phylogeny.
All unique sarbecovirus RBD protein-coding sequences were ordered from IDT, Twist or Genscript, and cloned into our yeast display vector26. Sequences were pooled and appended with downstream 16-nt barcode sequences according to the protocol described in Starr et al. 26. Long-read circular consensus sequences spanning the 16-nt barcode and RBD genotype were gathered on a PacBio Sequel v2.0 and processed exactly as described in Starr et al. 26. This yielded a barcode:variant lookup table for the sarbecovirus RBD library analogous to that used for SARS-CoV-2 mutant libraries. This table is available on GitHub (https://github.com/jbloomlab/SARSr-CoV_RBD_MAP/blob/main/data/barcode_variant_table.csv).
The pooled sarbecovirus RBD library was labelled, sorted, and quantified as described for the SARS-CoV-2 mutant libraries above, except we only sorted around 1 million RBD+ cells owing to the reduced library size. Sequencing and quantification of per-variant antibody escape was conducted as described above. Data for the HKU3-8 RBD are not shown, as this RBD was not successfully expressed in our yeast-display platform. For several antibodies, we performed a secondary experiment, selecting the sarbecovirus RBD library with a more stringent ‘full escape’ gate to select out only variants exhibiting complete loss of binding (Extended Data Fig. 2b, c).
For follow-up quantitative binding assays, select sarbecovirus RBDs were cloned into the yeast-display platform as isogenic stocks. Binding assays were conducted across a titration series of antibody in 96-well plates, and binding at each antibody concentration (geometric mean fluorescence intensity in the PE channel among RBD+ (FITC+) cells) was determined via flow cytometry and fit to a four-parameter Hill curve to identify the EC50 (midpoint).
Analysis of mutations in natural SARS-CoV-2 sequences
All spike sequences on GISAID59 as of 2 May 2021, were downloaded and aligned using mafft56. Sequences from non-human origins, sequences with gaps or ambiguous characters in the RBD, and sequences with more than eight amino acid differences from the Wuhan-Hu-1 reference sequence (GenBank MN908947, residues N331-T531) were removed. We determined mutation frequencies compared with Wuhan-Hu-1 reference from this final alignment of 1,190,241 sequences. We acknowledge all contributors to the GISAID EpiCoV database for their sharing of sequence data. The contributors to GISAID EpiCoV are listed at https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_Vir_mAbs/blob/main/data/gisaid_hcov-19_acknowledgement_table_2021_03_04.pdf.
Quantitative summary metrics of antibody properties
The relative epitope size of an antibody was calculated as the sum of per-mutant escape fractions that are at least five times the global median escape fraction (to minimize the impact of variation in background noise on the summation). For this summation, escape fractions were normalized to the maximum per-mutation escape fraction, to account for slight variation in the largest per-mutation escape fraction measured between selections.
The relative escapability of an antibody was calculated the same as relative epitope size, but each mutation was multiplied by two weighting factors scaled from 0 to 1 that reflect the impact of that mutation on ACE2-binding affinity and RBD expression as measured in our prior deep mutational scan26. The relationship between weighting factors and mutation effect on each property is shown in Extended Data Fig. 3a. Mutations with <–1 effect on either property are effectively zeroed out in the escapability summation. Mutations with effects between –1 and 0 have intermediate weights, and mutations with 0 or positive effects are assigned weight factors of 1.
Antibody susceptibility to escape by natural SARS-CoV-2 mutations was calculated as the summed GISAID frequencies of all escape mutations, where escape mutations (all labels in Extended Data Fig. 3c) are defined as those with escape fraction greater than five times the median escape fraction as above. These summed natural escape frequencies are tabulated in the plot headers in Extended Data Fig. 3c.
The summary breadth of an antibody was calculated from the sarbecovirus RBD library escape selection using the standard gating (Extended Data Fig. 4b), only. Although we have various follow-up binding data illustrating reduced affinity binding for some ‘escaped’ sarbecovirus RBDs, these follow-up experiments were not conducted systematically for all antibody–RBD combinations, and therefore would bias breadth estimates. Breadth of binding was calculated as the frequency of all sarbecovirus RBDs that are bound with affinity within the FACS selection gating threshold, weighted by clade representation. Breadth was normalized to give equal representation to each of the four sarbecovirus clades to account for different depth of sampling. Within the SARS-CoV-1 clade, all human 02/03 strains and civet and human 03/04 strains were similarly down-weighted to each represent 1/8 of the possible breadth within the SARS-CoV-1 clade (together with the six bat sarbecoviruses in this clade). As an example, breadth for S304 is calculated as [4/4 + ([6/6] + [6/6] + 5)/8 + 2/2 + 0/21]/4 = 0.72, based on the data shown in Extended Data Fig. 4b.
Multidimensional scaling projection of antibody epitopes
Multidimensional scaling projection in Fig. 4 was performed using the Python scikit-learn package. We first computed the similarity and dissimilarity in the sites of escape between each pair of antibodies, exactly as described in Greaney et al. 3, and performed metric multidimensional scaling with two components on the matrix of dissimilarities between all antibody pairs. Antibodies in this layout were coloured with pie charts proportional to the total squared site-wise escape that falls into the labelled structural regions (RBM, residues 437 to 508, ACE2 contact defined as 4 Å cut-off based on 6M0J crystal structure60, and core RBD otherwise). In this layout, we included all of our previously published antibodies for which we have performed escape mapping via this same approach. These antibodies and their citations include: S2X25937; LY-CoV55521; COV2-2196 and COV2-213036; REGN10933, REGN10987 and LY-CoV01620; and all other COV2 antibodies and CR30223.
For Fig. 4b–d and Extended Data Fig. 7c, we coloured the antibodies within this layout according to various antibody properties. When appropriate, we also coloured these previously assayed antibodies, as described below. Extended Data Fig. 7d and the scatter plots in Fig. 4e–g show the relationships between properties for antibodies specifically in this study (and S2X259) for the most direct comparability.
Antibody neutralization potencies illustrated in Fig. 4b incorporate the authentic SARS-CoV-2 neutralization IC50 values as reported in this study (Fig. 1a), together with the live SARS-CoV-2 neutralization IC50 values for the COV2 antibodies reported by Zost et al. 10. We acknowledge that it is imperfect to compare neutralization potencies reported from different labs on different antibody batches, though in this case, both sets are indeed neutralization potencies with authentic virus. We therefore do not directly compare these two sets of measurements in a quantitative manner, but we do note that their joint inclusion in Fig. 4b supports the dichotomy between neutralization potency of core RBD versus RBM antibodies which is supported by either neutralization panel alone.
Sarbecovirus breadth illustrated in Fig. 4c incorporates the pan-sarbecovirus breadth measurements reported in the current study together with more limited breadth measurements for antibodies reported in previous publications. These previously published experiments determined binding within a more restricted sarbecovirus RBD set present in our libraries (SARS-CoV-2, RaTG13, GD-pangolin, SARS-CoV-1 (Urbani), LYRa11 and WIV1). We calculated breadth from this incomplete sarbecovirus sequence set for comparison, but note that these antibodies are limited to a relative breadth of 0.5 because no RBDs from the Africa/Europe or non-ACE2-using Asia clades were included. However, as with neutralization, inclusion of these antibodies nonetheless emphasizes the core RBD–RBM dichotomy in sarbecovirus breadth established by our primary panel.
For illustrations of epitope size and escapability in Fig. 4d and Extended Data Fig. 7c, we calculated these quantities for our previously profiled antibodies as described above. We excluded the antibodies profiled in Greaney et al. 3, as these assays were performed on a prior version of our SARS-CoV-2 mutant library that exhibited different quantitative features of absolute escape, complicating its quantitative comparison to extent of escape for antibodies profiled in this and our other studies, which all use the same library.
Structural mappings around the perimeter of Fig. 4a were created by mapping total site-wise escape to the B-factor column of PDB structures. Footprints were defined as residues within a 5 Å cut-off of antibody heavy atoms. Structures used were those described in this paper, or previously published structures: ACE2-bound RBD (6M0J)60, CR3022-bound RBD (6W41)61, REGN10987- and REGN10933-bound RBD (6XDG)62, CB6- (LY-CoV016) bound RBD (7C01)63, and S304, S309 and S2H14-bound RBD (7JX3)15.
RBD ELISA
Ninety-six half-area well-plates (Corning, 3690) were coated over-night at 4 °C with 25 μl of sarbecoviruses RBD proteins at 5 μg ml−1 in PBS pH 7.2. Plates were blocked with PBS 1% BSA (Sigma-Aldrich, A3059) and subsequently incubated with serial dilutions of monoclonal antibody for 1 h at room temperature. After 4 washing steps with PBS 0.05% Tween 20 (PBS-T) (Sigma-Aldrich, 93773), goat anti-human IgG secondary antibody (Southern Biotech, 2040-04) was added and incubated for 1 h at room temperature. Plates were then washed 4 times with PBS-T and 4-nitrophenyl phosphate (pNPP, Sigma-Aldrich, 71768) substrate was added. After 30 min of incubation, absorbance at 405 nm was measured by a plate reader (Biotek) and data were plotted using GraphPad Prism.
Binding to cell surface-expressed sarbecovirus S proteins by flow cytometry
ExpiCHO-S cells were seeded at 6 × 106 cells per ml in a volume of 5 ml in a 50-ml bioreactor. Spike coding plasmids were diluted in cold OptiPRO SFM, mixed with ExpiFectamine CHO Reagent (Life Technologies) and added to the cells. Transfected cells were then incubated at 37 °C with 8% CO2 with an orbital shaking speed of 120 rpm (orbital diameter of 25 mm) for 42 h. Transiently transfected ExpiCHO-S cells were collected and washed two times in wash buffer (PBS, 1% BSA, 2 mM EDTA). Cells were counted and distributed into round bottom 96-well plates (Corning) and incubated with 10 μg ml−1 S2H97, S2X35 or S309 monoclonal antibody. Alexa Fluor647-labelled Goat Anti-Human IgG secondary Ab (Jackson ImmunoResearch 109-607-003) was prepared at 1.5 μg ml−1 and added to cells after two washing steps. Cells were then washed twice and resuspended in wash buffer for data acquisition on a ZE5 cytometer (Bio-Rad).
Crystallization, data collection, structure determination and analysis
To form RBD–Fab complexes for crystallization, SARS-CoV-2 RBD was mixed with a 1.3-fold molar excess of each Fab and incubated on ice for 20–60 min. Complexes were purified on a Superdex 200 Increase 10/300 GL column (Cytiva) preequilibrated with 20 mM Tris-HCl pH 7.5 and 150 mM NaCl. Crystals of the RBD–Fab complexes were obtained at 20 °C by sitting-drop vapour diffusion.
For the SARS-CoV-2 RBD–S2X35–S309 complex, a total of 200 nl complex at 5.4 mg ml−1 was mixed with 100 nl mother liquor solution containing 1.85 M ammonium sulfate, 0.1 M Tris pH 8.17, 0.8% (w/v) polyvinyl alcohol, 1% (v/v) 1-propanol, and 0.01 M HEPES pH 7. Crystals were flash frozen in liquid nitrogen using the mother liquor solution supplemented with 20% glycerol for cryoprotection. Data were collected at Beamline 9-2 of the Stanford Synchrotron Radiation Lightsource facility in Stanford, CA and processed with the XDS software package (version 31 January 2020)64 to 1.83 Å in space group C222. The RBD–S2X35–S309 Fab complex structure was solved by molecular replacement using phaser65 from a starting model consisting of SARS-CoV-2 RBD–S309 Fab (PDB 7JX3) and a homology model for the S2X35 Fab built using the Molecular Operating Environment (MOE) software package from the Chemical Computing Group (https://www.chemcomp.com).
For the SARS-CoV-2 RBD-S2H97-S2X259 Fab complex, 200 nl complex at 5.7 mg ml−1 was mixed with 200 nl mother liquor solution containing 0.12 M monosaccharides mix, 20% (v/v) ethylene glycol, 10% (w/v) PEG 8000, 0.1 M Tris (base)/bicine pH 8.5, 0.02 M sodium chloride, 0.01 M MES pH 6 and 3% (v/v) Jeffamine ED-2003. Crystals were flash frozen in liquid nitrogen. Data were collected at Beamline 9-2 of the Stanford Synchrotron Radiation Lightsource facility in Stanford, CA. Data were processed with the XDS software package (version Jan 31, 2020)64 for a final dataset of 2.65 Å in space group P21. The RBD–S2H97–S2X259 Fab complex structure was solved by molecular replacement using phaser from a starting model consisting of SARS-CoV-2 RBD (PDB 7JX3) and homology models for the S2H97 and S2X259 Fabs built using the MOE software package.
For the SARS-CoV-2 RBD–S2E12–S304–S309 Fab complex, 200 nl complex at 4.5 mg ml−1 was mixed with 100 nl of 0.09 M phosphate/citrate pH 5.5, 27% (v/v) PEG smear low, 4% (v/v) polypropylene glycol 400 and 0.02 M imidazole pH 7 or 100 nl of 0.09 M phosphate/citrate pH 5.5, 27% (v/v) PEG smear low, 0.01 M potassium/sodium phosphate pH 7, 1% (v/v) PPGBA 230 and 1.5% (v/v) PPGBA 400. Crystals were flash frozen in liquid nitrogen. Data were collected at the Molecular Biology Consortium beamline 4.2.2 at the Advanced Light Source synchrotron facility in Berkeley, CA. Datasets from two crystals from the two conditions were individually processed and then merged with the XDS software package (version 31 January 2020)64 for a final dataset of 2.93 Å in space group I4122. The RBD–S2E12–S304–S309 Fab complex structure was solved by molecular replacement using phaser from starting models consisting of SARS-CoV-2 RBD–S304–S309 Fab (PDB 7JX3) and S2E12 (PDB 7K3Q).
For all structures, several subsequent rounds of model building and refinement were performed using Coot (version 0.9.5)66, ISOLDE (ChimeraX version 1.1/ISOLDE version 1.1)67, Refmac5 (version 5.8.0267)68 and MOE (version 2019.0102) (https://www.chemcomp.com), to arrive at the final models. For all complexes, epitopes on the RBD protein were determined by identifying all RBD residues within a 5.0 Å distance from any Fab atoms. The analysis was performed using the MOE software package and the results were manually confirmed.
Cryo-electron microscopy
SARS-CoV-2 HexaPro S69 at 1.2 mg ml−1 was incubated with 1.2-fold molar excess of recombinantly purified S2D106 or S2H97 at 4 °C before application onto a freshly glow discharged 2.0/2.0 UltrAuFoil grid (200 mesh). Plunge freezing used a vitrobot MarkIV (Thermo Fisher Scientific) using a blot force of 0 and 6.5 s blot time at 100% humidity and 23 °C.
For the S/S2D106 dataset, data were acquired using an FEI Titan Krios transmission electron microscope operated at 300 kV and equipped with a Gatan K2 Summit direct detector and Gatan Quantum GIF energy filter, operated in zero-loss mode with a slit width of 20 eV. Automated data collection was carried out using Leginon70 at a nominal magnification of 130,000× with a pixel size of 0.525 Å. The dose rate was adjusted to 8 counts per pixel per s, and each movie was acquired in super-resolution mode fractionated in 50 frames of 200 ms. A total of 2,166 micrographs was collected, with a defocus range between −0.5 and −2.5 μm. Movie frame alignment, estimation of the microscope contrast-transfer function parameters, particle picking, and extraction were carried out using Warp71. Particle images were extracted with a box size of 800 binned to 400 pixels2 yielding a pixel size of 1.05 Å.
For the S/S2H97 dataset, data were acquired on an FEI Glacios transmission electron microscope operated at 200 kV equipped with a Gatan K2 Summit direct detector. Automated data collection was carried out using Leginon70 at a nominal magnification of 36,000× with a pixel size of 1.16 Å. The dose rate was adjusted to 8 counts per pixel per s, and each movie was acquired in counting mode fractionated in 50 frames of 200 ms. A total of 3,138 micrographs was collected in a single session with a defocus range comprised between −0.5 and −3.0 μm. Preprocessing was performed using Warp71 and particle images were extracted with a box size of 400 pixels2.
For the S/S2D106 and S/S2H97 datasets, two rounds of reference-free 2D classification were performed using CryoSPARC to select well-defined particle images72. These selected particles were subjected to two rounds of 3D classification with 50 iterations each (angular sampling 7.5° for 25 iterations and 1.8° with local search for 25 iterations), using our previously reported closed SARS-CoV-2 S structure as initial model49 (PDB 6VXX) in Relion73. 3D refinements were carried out using non-uniform refinement74 along with per-particle defocus refinement in CryoSPARC. Selected particle images were subjected to the Bayesian polishing procedure75 implemented in Relion3.0 before performing another round of non-uniform refinement in CryoSPARC followed by per-particle defocus refinement and again non-uniform refinement.
To further improve the density of the S2D106 Fab, the particles were then subjected to focus 3D classification without refining angles and shifts using a soft mask on RBD and Fab variable domains with a tau value of 60 in Relion. Particles belonging to classes with the best resolved local density were selected and subject to local refinement using CryoSPARC. Local resolution estimation, filtering, and sharpening were carried out using CryoSPARC. Reported resolutions are based on the gold-standard Fourier shell correlation (FSC) of 0.143 criterion and Fourier shell correlation curves were corrected for the effects of soft masking by high-resolution noise substitution76. UCSF Chimera77 and Coot78 were used to fit atomic models into the cryoEM maps. Spike-RBD/S2D106 Fab model was refined and relaxed using Rosetta using sharpened and unsharpened maps79.
S2H97-induced spike refolding
Ten micromolar native-like SARS-CoV-2 S was incubated with 13 μM S2H97 Fab for 1 h at room temperature. Samples were diluted to 0.01 mg ml−1 immediately before adsorption to glow-discharged carbon-coated copper grids for ~30 s before a 2% uranyl formate staining. Micrographs were recorded using the Leginon software70 on a 120 kV FEI Tecnai G2 Spirit with a Gatan Ultrascan 4000 4k × 4k CCD camera at 67,000× nominal magnification. The defocus ranged from −1.0 to −2.0 μm and the pixel size was 1.6 Å.
Cell-surface antibody-mediated S1 shedding
CHO-K1 cells stably expressing the prototypic SARS-CoV-2 spike protein were collected, washed in wash buffer (PBS, 1% BSA, 2 mM EDTA) and resuspended in PBS. Ninety-thousand cells per well were dispensed into round bottom 96-well plates (Corning), and treated with 10 μg ml−1 TPCK-Trypsin (Worthington Biochem) for 30 min at 37 °C. Cells were washed and incubated with 15 μg ml−1 antibody across 5, 30, 60, 120 and 180 min time points at 37 °C. Cells were washed with ice-cold wash buffer, and stained with 1.5 μg ml−1 Alexa Fluor647-conjugated goat anti-human IgG secondary antibody (Jackson Immunoresearch) for 30 min on ice in the dark. Cells were washed twice with cold wash buffer and analysed using a ZE5 cytometer (Bio-Rad) with acquisition chamber at 4 °C. Binding at each time point was measured as mean fluorescence intensity (MFI), normalized to the MFI at the 5 min labelling time point. Data were analysed and plotted using GraphPad Prism v. 9.0.1.
Cell-cell fusion of CHO-S cells
Cell–cell fusion between S-expressing CHO-K1 cells was performed as described by Lempp et al. 31. CHO-K1 cells stably expressing the prototypic SARS-CoV-2 spike protein were seeded in 96-well plates (Thermo Fisher Scientific) at 12,500 cells per well. The following day, antibody and nuclei marker Hoechst (final dilution 1:1,000) were added to cells and incubated for 24 h. Cell–cell fusion was visualized using the Cytation 5 Imager (BioTek), and an object detection protocol was used to detect nuclei and measure their size. The nuclei of fused cells (syncytia) are aggregated at the centre of the syncytia and recognized as a uniquely large object that is gated according to its size. To quantify cell–cell fusion, we report the area of objects in fused cells divided by the total area of all objects, multiplied by 100% to represent it as a percentage.
Antibody blockade of RBD binding to ACE2
ACE2 blockade ELISA was performed as described by Piccoli et al. 15. Unlabelled antibodies were serially diluted, mixed with RBD mouse Fc-tagged antigen (Sino Biological, final concentration 20 ng ml−1) and incubated for 30 min at 37 °C. The mix was added for 30 min to ELISA 96-well plates (Corning) pre-coated overnight at 4 °C with 2 μg ml−1 human ACE2 in PBS. Plates were washed and RBD binding was revealed using secondary goat anti-mouse IgG (Southern Biotech 1030-04). After washing, pNPP substrate was added and plates were read at 405 nm. The percentage of inhibition was calculated as: (1 – (ODsample – ODneg ctrl)/(ODpos ctrl – ODneg ctrl)) × 100%, where ODsample, ODneg ctrl and ODpos ctrl represent the optical densities of the sample, negative control and positive control, respectively.
Inhibition of spike-mediated cell–cell fusion
Cell–cell fusion inhibition assays were performed as described by McCallum et al. 80. Vero E6 cells were seeded in 96-well plates at 15,000 cells per well in 70 µl DMEM with high glucose and 2.4% FBS (Hyclone). After 16 h at 37 °C with 8% CO2, the cells were transfected as follows: for 10 wells, 0.57 µg plasmid SARS-CoV-2-S-D19_pcDNA3.1 was mixed with 1.68 µl X-tremeGENE HP in 30 µl OPTIMEM. After 15 min incubation, the mixture was diluted 1:10 in DMEM medium and 30 µl was added per well. Fourfold antibody serial dilutions were prepared and added to the cells, with a starting concentration of 20 μg ml−1. The following day, 30 μl 5×-concentrated DRAQ5 in DMEM was added per well and incubated for 2 h at 37 °C. Nine images of each well were acquired with a Cytation 5 equipment for analysis.
S2H97 prophylactic protection in Syrian hamsters
We used a validated SARS-CoV-2 Syrian golden hamster model of infection81,82 to test S2H97 prophylactic efficacy. Experiments were performed in the high-containment A3 and BSL3+ facilities of the KU Leuven Rega Institute (3CAPS) under licenses AMV 30112018 SBB 219 2018 0892 and AMV 23102017 SBB 219 20170589 according to institutional guidelines.
Syrian hamsters (Mesocricetus auratus) were purchased from Janvier Laboratories. Hamsters were housed per two in ventilated isolator cages (IsoCage N Biocontainment System, Tecniplast) with ad libitum access to food, water, and cage enrichment (wood block). Housing conditions and experimental procedures were approved by the ethical committee of animal experimentation of KU Leuven (license P065-2020). Sample sizes of 6 hamsters was determined in order to have a significant difference of at least 1 log(viral RNA level) (effect size d = 2.004) between control and treatment groups, by using a two-tailed t-test with 80% power and an alpha of 0.05, calculated with G*Power 3.1 software. Six- to ten-week-old female hamsters were randomized for administration of 25 mg kg−1 S2H97 antibody or 20 mg kg−1 human isotype control via intraperitoneal injection. Approximately 5 h before infection, animals were anesthetized with isoflurane to allow collection of a blood sample from the jugular vein to be used for antibody quantification. Forty-eight hours after antibody injection, hamsters were infected intranasally with 1.89 × 106 TCID50 SARS-CoV-2 virus in 50 μl inoculum. The challenge virus was a SARS-CoV-2 Wuhan isolate from February, 2020 (EPI_ISL_407976), passaged on Vero E6 cells. Passage 6 stock titre was determined by end-point dilution on Vero E6 cells by the Reed and Muench method83, expressed as 50% tissue culture infectious dose (TCID50).
Hamsters were monitored for appearance, behaviour, and weight. At day 4 post-infection, hamsters were euthanized by intraperitoneal injection of 500 μl Dolethal (200 mg ml−1 sodium pentobarbital, Vétoquinol SA). Lungs were collected, homogenized via bead disruption (Precellys) in 350 μl RLT buffer (RNeasy Mini kit, Qiagen) and centrifuged (10,000 rpm, 5 min, 4 °C) to pellet cell debris. RNA was extracted using a NucleoSpin kit (Macherey-Nagel) according to manufacturer instructions. Quantitative PCR with reverse transcription (RT–qPCR) was performed on a LightCycler96 platform (Roche) using the iTaq Universal Probes One-Step RT–qPCR kit (Bio-Rad) with N2 primers and probes targeting the nucleocapsid81. Standards of SARS-CoV-2 cDNA (IDT) were used to express viral genome copies per mg tissue. To quantify infectious SARS-CoV-2 particles, end-point titrations were performed on confluent Vero E6 cells in 96-well plates. Viral titres were calculated as above, and were expressed as TCID50 per mg tissue. The circulating antibody levels were measured by mesoscale bridging ELISA, using an anti-human LS mutation monoclonal antibody as a capture and anti-human CH2 monoclonal antibody as detection. Technicians performing RNA, virus, and antibody quantification were blinded to the treatment groups of processed samples. RNA and viral levels were compared between treatment and control via two-tailed Mann–Whitney test, excluding the two treatment animals with undetectable S2H97 levels at time of viral challenge.
Blockade of binding serology competition assays
Sera blockade of antibody binding was performed as described in Piccoli et al. 15. In brief, human IgG1 antibodies were biotinylated using the EZ-link NHS-PEO solid phase biotinylation kit (Pierce). Each labelled antibody was tested for binding to RBD by ELISA, and a concentration for each antibody competition experiment was selected to achieve 80% maximal binding (EC80). ELISA 96-well plates (Corning) were pre-coated overnight at 4 °C with 1 μg ml−1 of mouse Fc-tagged RBD antigen (Sino Biological) in PBS. Unlabelled sera or plasma were serially diluted and added to ELISA plates for 30 min, followed by addition of biotinylated anti-RBD antibody at its EC80 concentration. After 30 min incubation, plates were washed and antibody binding was detected using alkaline phosphatase-conjugated streptavidin (Jackson ImmunoResearch). Plates were washed, pNPP substrate (Sigma-Aldrich) was added, and plates were read at 405 nm. The percentage of inhibition of antibody binding was calculated as: (1–(ODsample – ODneg ctrl) / (ODpos ctrl – ODneg ctrl)) × 100.
Selection of VSV-SARS-CoV-2 monoclonal antibody resistance mutants
VSV-SARS-CoV-2 S chimera was used to select for SARS-CoV-2 S monoclonal antibody resistant mutants (MARMs) as previously described1,84. In brief, MARMs were recovered by plaque isolation on Vero E6 cells (ATCC, CRL-1586) with the indicated monoclonal antibody in the overlay. The concentration of monoclonal antibody in the overlay was determined by neutralization assays at a MOI of 100. Escape clones were plaque-purified on Vero cells (ATCC, CCL-81) in the presence of monoclonal antibody, and plaques in agarose plugs were amplified on MA104 cells (a gift from H. B. Greenberg (Stanford School of Medicine)) with the monoclonal antibody present in the medium. Viral stocks were amplified on MA104 cells at an MOI of 0.01 in Medium 199 containing 2% FBS and 20 mM HEPES pH 7.7 (Millipore Sigma) at 34 °C. Viral supernatants were collected upon extensive cytopathic effect and clarified of cell debris by centrifugation at 1,000g for 5 min. Aliquots were maintained at −80 °C. Viral RNA was extracted from VSV-SARS-CoV-2 mutant viruses using RNeasy Mini kit (Qiagen), and S was amplified using OneStep RT–PCR Kit (Qiagen). The mutations were identified by Sanger sequencing (Genewiz). Their resistance was verified by subsequent virus infection in the presence or absence of antibody. In brief, Vero cells were seeded into 12-well plates for overnight. The virus was serially diluted using DMEM and cells were infected at 37 °C for 1 h. Cells were cultured with an agarose overlay in the presence or absence of monoclonal antibody at 34 °C for 2 days. Plates were scanned on a biomolecular imager and expression of eGFP is shown at 48 h after infection. The S2X58-selected mutation S494L is not shown in Fig. 3a, as its effect on RBD expression was below the deep mutational scanning computational filter.
Viral replication fitness assays
Vero E6 cells (ATCC, CRL-1586) were seeded at 1 × 106 cells per well in 6-well plates. Cells were infected with MOI of 0.02, with wild-type and four mutant VSV-SARS-CoV-2 S chimeras mixed at equal (0.20) frequencies. Following 1 h incubation, cell monolayers were washed 3 times with HBBS and cultures were incubated for 72 h in humidified incubators at 34 °C. To passage the progeny viruses, virus mixture was continuously passaged 4 times in Vero E6 cells at MOI of 0.02. Cellular RNA samples from each passages were extracted using RNeasy Mini kit (QIAGEN) and subjected to next-generation sequencing as described previously to confirm the introduction and frequency of substitutions84.
Molecular dynamics simulations
Full details of molecular dynamics workflow and analysis are available on GitHub: https://github.com/choderalab/rbd-ab-contact-analysis. The RBD–S309 complex was constructed from PDB 7JX3 (chains A, B and R). 7JX3 was first refined using ISOLDE67. Refinement included adjusting several rotamers, flipping several peptide bonds, fixing several weakly resolved waters, and building in a missing four-residue-long loop. Though the N343 glycan N-Acetylglucosamine (NAG) was present in 7JX3, ISOLDE was used to construct a complex glycan at N343. The full glycosylation pattern was determined from refs. 85,86. The glycan structure used for N343 (FA2G2) corresponds to the most stable conformer obtained from multi microsecond molecular dynamics simulations of cumulative sampling87. The base NAG residue in FA2G2 was aligned to the corresponding NAG stub in the RBD-S309 model and any resulting clashes were refined in ISOLDE. The same process was repeated for the RBD–S2H97 crystal structure.
The refined glycosylated RBD–S309 and RBD–S2H97 complexes were prepared for simulation using tleap from AmberTools2088. All relevant disulfide bridges and covalent connections in glycan structures were specified. The glycosylated proteins were parameterized with the Amber ff14SB89 and GLYCAM_06j-190 force fields. The systems were solvated using the TIP3P rigid water model91 in a truncated octahedral box with 2.2 nm solvent padding on all sides. The shape and size of the solvent box were chosen to prevent the protein complex from interacting with its periodic image. The solvated systems were then neutralized with 0.15 M NaCl using the Li/Merz ion parameters of monovalent ions for the TIP3P water model (12-6 normal usage set)92. Virtual bonds were added across chains that should be imaged together to aid the post-processing of trajectories.
Each system was energy-minimized with an energy tolerance of 10 kJ mol−1 and equilibrated five times independently using the OpenMMTools 0.20.0 (https://github.com/choderalab/openmmtools) BAOAB Langevin integrator93 for 20 ns in the NPT (P = 1 atm, T = 310 K) ensemble with a time step of 4.0 fs, a collision rate of 1.0 ps−1, and a relative constraint tolerance of 1 × 10−5. Hydrogen atom masses were set to 4.0 amu by transferring mass from connected heavy atoms, bonds to hydrogen were constrained, and centre of mass motion was not removed. Pressure was controlled by a molecular-scaling Monte Carlo barostat with an update interval of 25 steps. Non-bonded interactions were treated with the Particle Mesh Ewald method94 using a real-space cut-off of 1.0 nm and the OpenMM default relative error tolerance of 0.0005, with grid spacing selected automatically. The simulations were subsequently packaged to seed for production simulation on Folding@home95,96. Default parameters were used unless noted otherwise.
The equilibrated structures (five per complex) were used to initiate parallel distributed molecular dynamics simulations on Folding@home95,96. Simulations were run with OpenMM 7.4.2 (compiled into Folding@home core22 0.0.13). Production simulations used the same Langevin integrator as the NPT equilibration described above. A total of 5,000 and 4,985 independent molecular dynamics simulations were generated on Folding@home for RBD–S309 and RBD–S2H97, respectively. Conformational snapshots (frames) were stored at an interval of 1 ns per frame for subsequent analysis. The final datasets contained 1.1 ms and 623.7 μs of aggregate simulation time for RBD–S309 and RBD–S2H97, respectively. This trajectory dataset (without solvent) are available at the MolSSI COVID-19 molecular structure and therapeutics hub (https://covid.molssi.org//simulations/#foldinghome-simulations-of-the-sars-cov-2-spike-rbd-bound-to-monoclonal-antibody-s309 and https://covid.molssi.org//simulations/#foldinghome-simulations-of-the-sars-cov-2-spike-rbd-bound-to-monoclonal-antibody-s2h97).
The first 100 ns of each trajectory was discarded (to allow relaxation away from the crystal structure), yielding total simulation times of 644.3 and 262.9 μs used for analysis of RBD–S309 and RBD–S2H97 systems, respectively. All trajectories had solute structures aligned to their first frame and centred using MDTraj97. Residues were considered to be at the interface if they were within 10 Å of any antibody Fab / RBD residue (with the exception of RBD N343 glycans, where all glycan residues were considered). The minimum distance of heavy atoms between every pair of interface residues was computed for every frame (1 ns) using MDAnalysis98,99. A close contact was counted if the minimum distance between a residue pair was below 3.5 Å (if one of the residues was hydrophobic, a 4.5 Å cut-off was used). The contribution of each RBD residue to close contacts was calculated as a percentage by summation of the number of close contacts for a particular RBD residue and normalizing by the total number of close contact interactions over all frames of each simulation.
Materials availability
The SARS-CoV-2 RBD mutant libraries (#1000000172) and unmutated parental plasmid (#166782) are available on Addgene. Other materials generated in this study will be made available on request and may require a material transfer agreement.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Interactive escape maps and structural visualizations can be found at https://jbloomlab.github.io/SARS-CoV-2-RBD_MAP_Vir_mAbs/. Raw Illumina sequencing data from deep mutational scanning experiments are available on NCBI Sequence Read Archive BioSample under accessions SAMN18315604 (SARS-CoV-2 mutant selection data) and SAMN18316011 (sarbecovirus RBD selection data). PacBio sequencing data used to link N16 barcodes to sarbecovirus RBD variant are available on NCBI SRA BioSample under accession SAMN18316101. A complete table of deep mutational scanning antibody escape fractions is provided on GitHub: https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_Vir_mAbs/blob/main/results/supp_data/all_antibodies_raw_data.csv. This table includes both antibodies first described in this study (Fig. 1b, c), and all other antibody selections that were re-processed to generate Fig. 4a. Coordinates and structure factors have been deposited in the Protein Data Bank (PDB) with accession code 7R6W for RBD-S2X35-S309, 7M7W for RBD-S2H97-S2X259 and 7R6X for RBD-S2E12-S304-S309. Cryo-electron microscopy structure data and models are available from the Electron Microscopy Data Bank (EMDB) under accession codes EMD-24300 for S/S2D106, EMDB EMD-24299 and PDB 7R7N for the S/S2D106 local refinement, and EMDB EMD-24301 for S/S2H97. The raw and processed molecular dynamics trajectory data are available at the MolSSI COVID-19 Molecular Structure and Therapeutics Hub: https://covid.molssi.org//simulations/#foldinghome-simulations-of-the-sars-cov-2-spike-rbd-bound-to-monoclonal-antibody-s309 and https://covid.molssi.org//simulations/#foldinghome-simulations-of-the-sars-cov-2-spike-rbd-bound-to-monoclonal-antibody-s2h97. All other datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request. Source data are provided with this paper.
Code availability
A repository containing all code, analysis and summary notebooks for the analysis of the SARS-CoV-2 deep mutational scanning escape selections is available on GitHub at https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_Vir_mAbs. A repository containing code and analysis of the sarbecovirus RBD library binding experiments is available on GitHub at https://github.com/jbloomlab/SARSr-CoV_RBD_MAP. A repository containing code and analysis of molecular dynamics simulations is available on GitHub at https://github.com/choderalab/rbd-ab-contact-analysis.
References
Liu, Z. et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 29, 477–488.e4 (2021).
Weisblum, Y. et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 9, e61312 (2020).
Greaney, A. J. et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 29, 44–57.e9 (2021).
Pinto, D. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295 (2020).
Rappazzo, C. G. et al. Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody. Science 371, 823–829 (2021).
Menachery, V. D. et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 21, 1508–1513 (2015).
Letko, M., Marzi, A. & Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 5, 562–569 (2020).
Tortorici, M. A. et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science 370, 950–957 (2020).
Barnes, C. O. et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 588, 682–687 (2020).
Zost, S. J. et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 584, 443–449 (2020).
Hassan, A. O. et al. A SARS-CoV-2 infection model in mice demonstrates protection by neutralizing antibodies. Cell 182, 744–753.e4 (2020).
Winkler, E. S. et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell 184, 1804–1820.e16 (2021).
Schäfer, A. et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J. Exp. Med. 218, e20201993 (2021).
Corti, D., Purcell, L. A., Snell, G. & Veesler, D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell 184, 3086–3108 (2021).
Piccoli, L. et al. Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell 183, 1024–1042.e21 (2020).
Greaney, A. J. et al. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 29, 463–476.e6 (2021).
Chen, R. E. et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 27, 717–726 (2021).
Wang, P. et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 593, 130–135 (2021).
Thomson, E. C. et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 184, 1171–1187.e20 (2021).
Starr, T. N. et al. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 371, 850–854 (2021).
Starr, T. N., Greaney, A. J., Dingens, A. S. & Bloom, J. D. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep. Med. 2, 100255 (2021).
Cathcart, A. L. et al. The dual function monoclonal antibodies VIR-7831 and VIR-7832 demonstrate potent in vitro and in vivo activity against SARS-CoV-2. Preprint at https://doi.org/10.1101/2021.03.09.434607 (2021).
McCallum, M. et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science https://doi.org/10.1126/science.abi7994 (2021).
Gupta, A. et al. Early Covid-19 treatment with SARS-CoV-2 neutralizing antibody sotrovimab. Preprint at https://doi.org/10.1101/2021.05.27.21257096 (2021).
Dingens, A. S., Arenz, D., Weight, H., Overbaugh, J. & Bloom, J. D. An antigenic atlas of HIV-1 escape from broadly neutralizing antibodies distinguishes functional and structural epitopes. Immunity 50, 520–532.e3 (2019).
Starr, T. N. et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 182, 1295–1310.e20 (2020).
Faria, N. R. et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 372, 815–821 (2021).
Tegally, H. et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 592, 438–443 (2021).
Zhang, W. et al. Emergence of a novel SARS-CoV-2 variant in southern California. J. Am. Med. Assoc. 325, 1324–1326 (2021).
Walls, A. C. et al. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell 176, 1026–1039.e15 (2019).
Lempp, F. A. et al. Membrane lectins enhance SARS-CoV-2 infection and influence the neutralizing activity of different classes of antibodies. Preprint at https://doi.org/10.1101/2021.04.03.438258 (2021).
Pinto, D. et al. A human antibody that broadly neutralizes betacoronaviruses protects against SARS-CoV-2 by blocking the fusion machinery. Preprint at https://doi.org/10.1101/2021.05.09.442808 (2021).
Walls, A. C. et al. Elicitation of potent neutralizing antibody responses by designed protein nanoparticle vaccines for SARS-CoV-2. Cell 183, 1367–1382.e17 (2020).
Cohen, A. A. et al. Mosaic nanoparticles elicit cross-reactive immune responses to zoonotic coronaviruses in mice. Science 371, 735–741 (2021).
Saunders, K. O. et al. Neutralizing antibody vaccine for pandemic and pre-emergent coronaviruses. Nature 594, 553–559 (2021).
Dong, J. et al. Genetic and structural basis for recognition of SARS-CoV-2 spike protein by a two-antibody cocktail. Preprint at https://doi.org/10.1101/2021.01.27.428529 (2021).
Tortorici, M. A. et al. Broad sarbecovirus neutralization by a human monoclonal antibody. Nature https://doi.org/10.1038/s41586-021-03817-4 (2021).
Sauer, M. M. et al. Structural basis for broad coronavirus neutralization. Nat. Struct. Mol. Biol. 28, 478–486 (2021).
Kistler, K. E. & Bedford, T. Evidence for adaptive evolution in the receptor-binding domain of seasonal coronaviruses OC43 and 229e. eLife 10, e64509 (2021).
Eguia, R. T. et al. A human coronavirus evolves antigenically to escape antibody immunity. PLoS Pathog. 17, e1009453 (2021).
Walls, A. C. et al. Elicitation of broadly protective sarbecovirus immunity by receptor-binding domain nanoparticle vaccines. Preprint at https://doi.org/10.1101/2021.03.15.435528 (2021).
Zhou, P. et al. A protective broadly cross-reactive human antibody defines a conserved site of vulnerability on beta-coronavirus spikes. Preprint at https://doi.org/10.1101/2021.03.30.437769 (2021).
Crawford, K. H. D. et al. Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 spike protein for neutralization assays. Viruses 12, 513 (2020).
Xie, X. et al. A nanoluciferase SARS-CoV-2 for rapid neutralization testing and screening of anti-infective drugs for COVID-19. Nat. Commun. 11, 5214 (2020).
Takada, A. et al. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl Acad. Sci. USA 94, 14764–14769 (1997).
Riblett, A. M. et al. A haploid genetic screen identifies heparan sulfate proteoglycans supporting rift valley fever virus infection. J. Virol. 90, 1414–1423 (2015).
Giroglou, T. et al. Retroviral vectors pseudotyped with severe acute respiratory syndrome coronavirus S protein. J. Virol. 78, 9007–9015 (2004).
Collier, D. A. et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 593, 136–141 (2021).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e6 (2020).
Otwinowski, J., McCandlish, D. M. & Plotkin, J. B. Inferring the shape of global epistasis. Proc. Natl Acad. Sci. USA 115, E7550–E7558 (2018).
Hilton, S. K. et al. dms-view: Interactive visualization tool for deep mutational scanning data. J. Open Source Softw. 5, 2353 (2020).
Song, H.-D. et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl Acad. Sci. USA 102, 2430–2435 (2005).
Tong, S. et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 15, 482–485 (2009).
Zhou, H. et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 30, 2196–2203.e3 (2020).
Lam, T. T.-Y. et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583, 282–285 (2020).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 (2006).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Elbe, S. & Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 1, 33–46 (2017).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Yuan, M. et al. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 368, 630–633 (2020).
Hansen, J. et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 369, 1010–1014 (2020).
Shi, R. et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 584, 120–124 (2020).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D 74, 519–530 (2018).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367 (2011).
Hsieh, C.-L. et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 369, 1501–1505 (2020).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Tegunov, D. & Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Zivanov, J., Nakane, T. & Scheres, S. H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019).
Ramlaul, K., Palmer, C. M., Nakane, T. & Aylett, C. H. S. Mitigating local over-fitting during single particle reconstruction with SIDESPLITTER. J. Struct. Biol. 211, 107545 (2020).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Casañal, A., Lohkamp, B. & Emsley, P. Current developments in Coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci. 29, 1069–1078 (2020).
Wang, R. Y.-R. et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife 5, e17219 (2016).
McCallum, M. et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 184, 2332–2347.e16 (2021).
Boudewijns, R. et al. STAT2 signaling restricts viral dissemination but drives severe pneumonia in SARS-CoV-2 infected hamsters. Nat. Commun. 11, 5838 (2020).
Sanchez-Felipe, L. et al. A single-dose live-attenuated YF17D-vectored SARS-CoV-2 vaccine candidate. Nature 590, 320–325 (2021).
Reed, L. J. & Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 27, 493–497 (1938).
Case, J. B. et al. Neutralizing antibody and soluble ACE2 inhibition of a replication-competent VSV-SARS-CoV-2 and a clinical isolate of SARS-CoV-2. Cell Host Microbe 28, 475–485.e5 (2020).
Shajahan, A. et al. Comprehensive characterization of N- and O-glycosylation of SARS-CoV-2 human receptor angiotensin converting enzyme 2. Glycobiology 31, 410–424 (2020).
Watanabe, Y., Allen, J. D., Wrapp, D., McLellan, J. S. & Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 369, 330–333 (2020).
Harbison, A. M., Brosnan, L. P., Fenlon, K. & Fadda, E. Sequence-to-structure dependence of isolated IgG Fc complex biantennary N-glycans: a molecular dynamics study. Glycobiology 29, 94–103 (2019).
Case, D.A. et al. Amber 2020 (Univ. of California, 2020).
Maier, J. A. et al. Ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Kirschner, K. N. et al. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 29, 622–655 (2008).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Li, P., Song, L. F. & Merz, K. M., Jr Systematic parameterization of monovalent ions employing the nonbonded model. J. Chem. Theory Comput. 11, 1645–1657 (2015).
Leimkuhler, B. & Matthews, C. Robust and efficient configurational molecular sampling via Langevin dynamics. J. Chem. Phys. 138, 174102 (2013).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Shirts, M. & Pande, V. S. COMPUTING: Screen savers of the world unite! Science 290, 1903–1904 (2000).
Zimmerman, M. I. et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 13, 651–659 (2021).
McGibbon, R. T. et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 (2015).
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B. & Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327 (2011).
Gowers, R. et al. MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. in Proc. 15th Python in Science Conference (SciPy, 2016).
Acknowledgements
We thank I. Hoffman for assistance in refinement of crystal structures, G. R. Bowman, J. Coffland and P. K. Eastman for developing and maintaining the Folding@home infrastructure, Amazon Web Services for Folding@home infrastructure support, the Folding@home volunteers who contributed their computational resources to this project (FAH Project 17336-17340), R. P. Wiewiora and S. Singh for their assistance with Folding@home, and A. M. Harbison and E. Fadda for assistance in glycan modeling. This work was supported by the Fred Hutch Flow Cytometry and Genomics facilities, the Fred Hutch Scientific Computing group supported by ORIP grant S10OD028685, and the University of Washington Arnold and Mabel Beckman Cryo-EM Center. This work was supported by the NIH/NIAID (R01AI127893 and R01AI141707 to J.D.B., DP1AI158186 and HHSN272201700059C to D.V. and T32AI083203 to A.J.G.), the NIH/NIGMS (R01GM120553 to D.V., and grant R01GM121505 and R01GM132386 to J.D.C.), the NIH/NCI (P30CA008748 to J.D.C.), the National Science Foundation (NSF CHI-1904822 to J.D.C.), the Damon Runyon Cancer Research Foundation (T.N.S.), the Gates Foundation (INV-004949 to J.D.B.), a Pew Biomedical Scholars Award (D.V.), Investigators in the Pathogenesis of Infectious Disease Awards from the Burroughs Wellcome Fund (J.D.B. and D.V.), the Wellcome Trust (209407/Z/17/Z to T.I.C.), Fast Grants (D.V.), Bayer (W.G.G.) and the Molecular Sciences Software Institute (I.Z.). J.D.B. is an Investigator of the Howard Hughes Medical Institute. The Molecular Biology Consortium beamline 4.2.2 of the Advanced Light Source, a US DOE Office of Science User Facility under contract no. DE-AC02-05CH11231, is supported in part by the ALS-ENABLE program funded by the NIH/NIGMS (P30GM124169-01). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the NIH/NIGMS (P30GM133894).
Author information
Authors and Affiliations
Contributions
Conceived research and designed study: T.N.S., N.C., H.W.V., D.C., J.D.B. and G.S. Antibody discovery: F.Z., D.P., M.B., R.M., A.D.M., E.C., M.S.P. and D.C. Expression and purification of proteins: N.C., P.H., G.L., N.S., J.E.B., A.C.W., K.C., S.J. and M.M. Antibody functional experiments: Y.-J.P., Z.L., F.Z., D.P., M.B., R.M., J.E.B., M.A.T., A.C.W., J.A.W., A.D.M., L.E.R., J.Z., M.M.-R., H.K., J.R.D., H.T., J.B., C.S.-F., M.P.H., M.A., E.D.Jr, S.S., C.H.-D., L.P., F.B., F.A.L. and S.P.J.W. Deep mutational scanning experiments and analysis: T.N.S., A.A., A.J.G. and A.S.D. Hamster model: R.A., S.-Y.C.F., F.B. and J.N. Bioinformatics analysis: J.d.I. and A.T. Structure determination: N.C., Y.-J.P., P.H., J.E.B., T.I.C., J.C.N., D.V. and G.S. Molecular dynamics simulation and analysis: W.G.G., I.Z. and J.D.C. Supervision: M.S.P., J.D.C., C.M.H., S.P.J.W., D.V., D.C., J.D.B. and G.S. Writing of the initial draft: T.N.S., N.C., D.C., J.D.B. and G.S. Editing of the final version: all authors.
Corresponding authors
Ethics declarations
Competing interests
N.C., F.Z., D.P., M.B., P.H., R.M., J.A.W., A.D.M., L.E.R., J.Z., M.M.-R., H.K., J.R.D., H.T., J.B., C.S.-F., M.P.H., J.d.I., G.L., M.A., N.S., K.C., S.J., M.M., E.D.Jr, E.C., C.H.-D., L.P., F.B., A.T., F.A.L., M.S.P., C.M.H., H.W.V., D.C. and G.S. are or were employees of Vir Biotechnology and may hold shares in Vir Biotechnology. D.C. is currently listed as an inventor on multiple patent applications, which disclose the subject matter described in this manuscript. After the submission of the initial version of this study, J.D.B. began consulting for Moderna on viral evolution and epidemiology. J.D.B. has the potential to receive a share of IP revenue as an inventor on a Fred Hutchinson Cancer Research Center-optioned technology/patent (application WO2020006494) related to deep mutational scanning of viral proteins. H.W.V. is a founder of PierianDx and Casma Therapeutics. Neither company provided funding for this work nor is performing related work. J.C.N., T.I.C. and D.V. are consultants for Vir Biotechnology Inc. The Veesler laboratory has received a sponsored research agreement from Vir Biotechnology Inc. J.D.C. is a current member of the Scientific Advisory Boards of OpenEye Scientific Software, Interline Therapeutics, and Redesign Science. The Chodera laboratory receives or has received funding from the National Institute of Health, the National Science Foundation, the Parker Institute for Cancer Immunotherapy, Relay Therapeutics, Entasis Therapeutics, Silicon Therapeutics, EMD Serono (Merck KGaA), AstraZeneca, Vir Biotechnology, XtalPi, Interline Therapeutics, and the Molecular Sciences Software Institute, the Starr Cancer Consortium, the Open Force Field Consortium, Cycle for Survival, a Louis V. Gerstner Young Investigator Award, and the Sloan Kettering Institute. A complete funding history for the Chodera lab can be found at http://choderalab.org/funding. The other authors declare no competing interests.
Additional information
Peer review information Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Antibody neutralization and binding data.
a, Neutralization of authentic SARS-CoV-2 (SARS-CoV-2-Nluc) by 14 antibodies. Shown are representative live virus neutralization plots, measured with entry into Vero E6 cells. Symbols are means ± s.d. of technical triplicates. Dashed lines indicate IC50 and IC90 values. All antibodies were measured at each concentration point in the series, with hidden points due to overplotting reflecting overlap at the upper and lower neutralization limits. b, Correlation in antibody neutralization IC50 as determined in spike-pseudotyped VSV particles (n = 3 to 8) versus authentic SARS-CoV-2 (n = 3). c, Representative SPR sensorgrams of Fab fragments of the six newly described antibodies binding to the SARS-CoV-2 RBD (Fig.1a gives average values for n = 2–4 measurements). White and grey stripes indicate association and dissociation phases, respectively. Data shown in blue and fit to binding model shown in dashed gray line. Binding affinities for previously described antibodies shown in Fig. 1a are consistent with measurements from Piccoli et al. (S304, S309, S2X35, S2H13, S2H14)15 and Tortorici et al. (S2E12)8. d, Identifiers and spike genotypes of SARS-CoV-2 variants tested in neutralization assays in Figs. 2d, 3b.
Extended Data Fig. 2 Deep mutational scanning to map mutations that escape antibody binding.
a, Scheme of the deep mutational scanning assay. Conformationally intact RBD is expressed on the surface of yeast, where RBD expression and antibody binding is detectable via fluorescent labelling. We previously constructed mutant libraries containing virtually all of the 3,819 possible amino acid mutations in the SARS-CoV-2 RBD26 and sorted the library to eliminate mutations that destabilize the RBD or strongly reduce ACE2-binding affinity3. We incubate the library with a sub-saturating antibody concentration and use fluorescence-activated cell sorting (FACS) to isolate yeast cells expressing RBD mutants with reduced antibody binding. Deep sequencing quantifies mutant frequencies before and after FACS selection, enabling calculation of the “escape fraction” of each amino acid mutation, which reflects the fraction of cells carrying that mutation that fall into the antibody-escape bin. Mutation escape fractions are represented in logo plots, where the height of a letter reflects the extent of escape from antibody binding. b, Representative selection gates, after gating for single cells expressing RBD as in Greaney et al. 3. Yeast expressing the SARS-CoV-2 RBD (top panels) are labelled at 1x, 0.01x and no antibody to guide selection gates. Mutant RBDs that reduce binding (green, gate drawn to capture 0.01x wild-type control) are sorted and sequenced for calculation of mutant escape fractions. This same gate was used to quantify escape within libraries of yeast expressing all sarbecovirus RBD homologues. For several antibodies, we also selected the sarbecovirus RBD library with a more stringent “full escape” gate (blue, gate drawn to capture 0 ng ml−1 wild-type control). c, Fraction of library cells falling into escape bins for each antibody selection. d, Line plots showing total escape at all RBD sites for each antibody. Sites of strong escape illustrated in logo plots in Fig. 1b, c shown with pink indicators. e, f, Correlation in per-mutation (e) and per-site (f, sum of per-mutation) escape fractions for duplicate libraries that were independently generated and assayed. N, number of mutations (e) or sites (f) in the correlation.
Extended Data Fig. 3 Antibody escapability from deep mutational scanning measurements and in natural SARS-CoV-2 mutants.
a, To calculate antibody escapability (Fig. 1b, c), mutation escape fractions were weighted by their deleterious consequences for ACE2 binding or RBD expression. Top plots show the weighting factor (y-axis) for mutation effects on ACE2 binding (left) and RBD expression (right). This weight factor was multiplied by the mutation escape fraction in the summation to calculate antibody escapability as described in the Methods. Histograms show the distribution of mutation effects on ACE2 binding (left) and RBD expression (right) for all mutations that pass our computational filtering steps (bottom), and mutations that are found with at least 20 sequence counts on GISAID (middle). b, Correlation in antibody relative epitope size (top) and escapability (bottom) calculated from independent deep mutational scanning replicates, compared to the averaged replicates shown in Fig. 1b, c. R2, squared Pearson correlation coefficient. c, Scatter plots illustrate the degree to which a mutation escapes antibody binding (escape fraction, y-axis) and its frequency among 1,190,241 high-quality human-derived SARS-CoV-2 sequences present on GISAID as of May 2, 2021. Large escape mutations (>5x global median escape fraction) for each antibody with non-zero mutant frequencies are labelled. Plot labels report the sum of mutant frequencies for all labelled mutations, corresponding to the natural SARS-CoV-2 mutant escape frequency for antibodies shown in Fig. 4d, g.
Extended Data Fig. 4 Breadth of antibody binding across sarbecoviruses.
a, Phylogenetic relationship of sarbecovirus RBDs inferred from aligned nucleotide sequences, with the four sarbecovirus clades labelled in separate colours used throughout the text. Node support values are bootstrap support values. b, Breadth of sarbecovirus binding by each antibody to a panel of yeast-displayed sarbecovirus RBDs. Data as in Fig. 1d, with the addition of secondary “full escape” selection data for S2H97, S2H13, and S2H14 (0 ng ml−1 wild-type control as shown in Extended Data Fig. 2b, c), enabling differentiation of RBDs with intermediate binding (for example, S2H97/RsSHC014) versus complete loss of binding. Escape fractions are calculated as the mean of replicate barcoded genotypes internal to the library. Median number of barcodes per RBD is 249, with a range of 104 to 566. The median SEM across escape fraction measurements is 0.019, with a range of 0.00005 to 0.038 across all RBD/antibody pairs. c, Flow cytometry detection of antibody binding to isogenic yeast-displayed RBD variants. d, Flow cytometry detection of antibody binding to mammalian-surface displayed spikes. e, ELISA binding of antibody to purified RBD proteins. f, SPR measurement of binding of cross-reactive antibodies (Fab) and human ACE2 to select sarbecovirus RBDs. NB, no binding; NT, not tested. Values from single replicates. g, S2H97 neutralization of VSV pseudotyped with select sarbecovirus spikes, with entry measured in ACE2-transduced BHK-21 cells. Curves are representative of two independent experiments. Points represent means, error bars standard deviation from three technical replicates, and IC50 geometric mean of experiments. IC50 values are not comparable to other experiments on Vero E6 cells (for example, Fig. 2c) due to ACE2 overexpression and its impact on S2H97 neutralization. h, Alignment of germline-reverted and mature S2H97 heavy- (top) and light-chain (bottom) amino acid sequences. CDR sequences shown in grey box. Heat map overlay indicates the predicted energetic contribution of antibody paratope residues from the crystal structure. i, Binding of germline-reverted and mature S2H97 to select sarbecovirus RBDs as measured by SPR. j, Neutralization experiment of select sarbecoviruses by S2E12 (spike-pseudotyped VSV on 293T-ACE2 cells). Details as in Fig. 3c. k, Alignment of germline-reverted and mature S2E12. Details as in h. l, Binding of germline-reverted and mature S2E12 to select sarbecovirus RBDs as measured by SPR.
Extended Data Fig. 5 Structures and epitopes of Fab:RBD complexes.
a, Surfaces targeted by broadly binding RBD antibodies. RBD surface is coloured by site variability across sarbecoviruses. ACE2 key motifs shown in transparent red cartoon. Antibody variable domains shown as cartoon, with darker shade indicating the heavy chain. b, c, Integrative features of the S309 (b) and S2X35 (c) structural epitopes. Details as in Figs. 3g, h, 2b. Variants in the S309 epitope were tested with VIR-7831 with the exception of E340D (marked with *), which was tested with S309. d–h, Zoomed in view of the RBD bound to S309 (d), S2X35 (e), S2H97 (f), S2E12 (g), and S2D106 (h), with important contact and escape residues labelled. RBD residues coloured by total site escape [scale bar, right of (d)]. i, j, Representative electron micrograph and 2D class averages of SARS-CoV-2 S in complex with the S2H97 Fab embedded in vitreous ice. Scale bar: 400 Å. Micrographs representative of 3138 micrographs. k, Gold-standard Fourier shell correlation curve for the S2H97-bound SARS-CoV-2 S trimer reconstruction. The 0.143 cut-off is indicated by a horizontal dashed line. l, Local resolution map calculated using cryoSPARC for the whole reconstruction with two orthogonal orientations. m, n, Representative electron micrograph and 2D class averages of SARS-CoV-2 S in complex with the S2D106 Fab embedded in vitreous ice. Scale bar: 400 Å. Micrographs representative of 2166 micrographs o, Gold-standard Fourier shell correlation curves for the S2D106-bound SARS-CoV-2 S trimer (black line) and locally refined RBD/S2D106 variable domains (grey line). The 0.143 cut-off is indicated by a horizontal dashed line. p, Local resolution map calculated using cryoSPARC for the whole reconstruction and the locally refined RBD/S2D106 variable domain region.
Extended Data Fig. 6 Mechanism of action of S2H97 neutralization and protection.
a, Quaternary context of the S2H97 epitope. Left, S2H97-bound RBD, with RBD sites coloured by S2H97 escape (scale bar, bottom). Right, RBD in the same angle as left, in the closed spike trimer. b, CryoEM structure of S2H97 Fabs (green surfaces) bound to SARS-CoV-2 S indicating the extensive opening of the RBD (yellow surface) necessary to access the S2H97 epitope. Closed RBD (light purple surface, PDB 7K43) and site II Fab S2A4 bound open RBD (grey surface, PDB 7JVC) are shown for comparison. Spike protomers are shown in yellow, blue, and pink. c, Antibody-mediated S1 shedding from cell-surface expressed SARS-CoV-2 S as determined by flow cytometry. d, Cell-cell fusion of CHO cells expressing SARS-CoV-2 S (CHO-S) incubated with variable concentrations of antibody. e, Antibody competition with RBD-ACE2 binding determined by ELISA. Points represent mean of technical duplicates. f, S2H97 neutralization of SARS-CoV-2 S pseudotyped VSV on ACE2-overexpressing cells (293T-ACE2) compared to Vero E6 cells. Points reflect mean and error bars reflect standard deviation from triplicate measurements. Curves are representative of two biological replicates. g, Antibody inhibition of cell-to-cell fusion of Vero E6 cells transfected with SARS-CoV-2 S. h, Influence of circulating S2H97 level on prophylactic efficacy in Syrian hamsters. Infectious virus titres (right y-axis, triangles) and RNA levels (left y-axis, circles) reflect the data represented in Fig. 2f, measured in hamsters four days after SARS-CoV-2 challenge in animals prophylactically dosed with 25 mg/kg S2H97 (magenta symbols) or isotype control (white symbols). The levels of circulating S2H97 (D0, before infection, μg ml−1) are shown on the x-axis (LLOQ, lower level of quantification). ** P = 0.0048 (virus titre) and P = 0.0048 (RNA) vs control isotype, two-sided Mann–Whitney test (the 2 animals shown with no detectable serum antibody were excluded from the comparison).
Extended Data Fig. 7 Escapability and the relationships among antibody properties.
a, Additional spike-VSV viral escape selections, as in Fig. 3a, and an illustration of the authentic SARS-CoV-2 escape data for VIR-7832 (derived from S309) reported in Cathcart et al. 22. b, Correlation between the number of unique mutations selected across viral escape selection experiments and antibody escapability as tabulated in Fig. 1b, c, plus S2X25937. c, Projected epitope space from Fig. 4a annotated by antibody properties as in Fig. 4b–d. For each property, antibodies are coloured such that purple reflects the most desirable antibody (scale bar, right; N.D., not determined): narrowest functional epitope, tightest binding affinity (KD, log10 scale), lowest escapability. d, Pairwise scatter plots between all antibody properties discussed in the main text. Select scatter plots from this panel are shown in Figs. 4e–g. Details of each property described in Methods. All axes are oriented such that moving up on the y-axis and right on the x-axis corresponds to moving in the “preferred” direction for an antibody property (lower neutralization IC50, lower KD, higher breadth, narrower epitope size, lower escapability, lower total frequency of SARS-CoV-2 escape mutants among sequences on GISAID).
Supplementary information
Source data
Rights and permissions
About this article

Cite this article
Starr, T.N., Czudnochowski, N., Liu, Z. et al. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature 597, 97–102 (2021). https://doi.org/10.1038/s41586-021-03807-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03807-6
This article is cited by
-
Potent and broadly neutralizing antibodies against sarbecoviruses induced by sequential COVID-19 vaccination
Cell Discovery (2024)
-
Repeated Omicron exposures override ancestral SARS-CoV-2 immune imprinting
Nature (2024)
-
A human antibody derived from original SARS-CoV-2 infection effectively neutralizes omicron
Advanced Biotechnology (2024)
-
Identification of a highly conserved neutralizing epitope within the RBD region of diverse SARS-CoV-2 variants
Nature Communications (2024)
-
A monoclonal antibody targeting a large surface of the receptor binding motif shows pan-neutralizing SARS-CoV-2 activity
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
