
Abstract
This study aims to review the available literature pertinent to vascular complications in COVID-19. A systematic search was performed using PubMed and Google Scholar to identify all relevant studies based on our study objective. Multiple studies have reported widespread systemic inflammation and procoagulant/hypercoagulable state in COVID-19, including thrombotic microangiopathy, endothelial dysfunction, bleeding disorder, and thrombosis. However, large specialised studies on vascular complications are lacking despite current evidence indicating dysfunctional coagulation pathways. Furthermore, there are no clear and definitive recommendations regarding thromboprophylaxis or full therapeutic anticoagulation in COVID-19. Several studies have reported hypercoagulability and vascular complications as important predictors of patient outcome in COVID-19. Therefore, it is important to understand the pathogenesis, epidemiology, management, and outcomes of patients who develop venous or arterial thrombosis and those with a pre-existing thrombotic disease who contract COVID-19 for risk stratification, thromboprophylaxis, optimal antithrombotic therapy during active infection and long-term anticoagulation following discharge or recovery.
Similar content being viewed by others
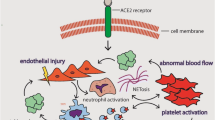

Highlights
-
SARS-CoV-2 induces proinflammatory cytokines and/or procoagulant factors that could activate coagulation cascades, leading to thrombosis, atherosclerotic plaque rupture, and ischemia.
-
Noting changes in coagulation profile in critically ill COVID-19 patients are of utmost importance, as studies have shown that hypercoagulability and vascular complications are important predictors of optimal patient outcomes.
-
Despite current evidence indicating dysfunctional coagulation pathways, specific studies concerning vascular complications in COVID-19 are lacking.
-
It is crucial to have large-scale multicentre studies to establish and ascertain the vascular complications in COVID-19 and formulate management strategies.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes an aggressive infection, coronavirus disease 2019 (COVID-19), with potentially serious health consequences [1]. Although COVID-19 primarily causes respiratory infection, recent studies have shifted the attention to vascular complications, including alarming thromboembolism [2,3]. Studies have shown alterations in the coagulation pathway such as elevated D-dimer levels, fibrin breakdown, reduced anti-thrombin levels, and reduced prothrombin and thrombin time [4,5,6,7,8,9,10]. Noting changes in coagulation profile in critically ill patients is of utmost importance as several studies have reported hypercoagulability and vascular complications as important predictors of patient outcome. As new evidence is unfolding, we aim to review the available literature pertinent to the vascular complications in COVID-19.
Methods
A literature review was performed using PubMed and Google Scholar to identify all relevant studies based on our study objective. Non-specific combinations of the search strings included, (i) Coronavirus OR Severe acute respiratory syndrome OR 2019-nCoV OR SARS-CoV-2 OR Severe acute respiratory syndrome coronavirus 2 OR Coronavirus disease 2019 OR COVID-19, (ii) Vascular complications OR Vascular injury OR Vascular pathology OR Vasculitis (iii) Endothelial dysfunction, (iv) Thrombosis OR Thromboembolism, and (v) Coagulation dysfunction OR Coagulopathy. Only the studies pertinent to vascular complications reported within the background of the COVID-19 infection were included. Independent analysis of all the studies was performed due to the unavailability of randomised clinical trials (RCT). A secondary reference search was conducted to find any additional studies. In total, we included 80 studies amongst the 291 studies that we analysed based on the study objective and search strategy.
Emergence of evidence
During the early stage of the COVID-19 pandemic, Zhang et al. [11] compared the clinical and pathologic features of the severe acute respiratory syndrome (SARS) and SARS-CoV-2. Diffuse alveolar damage (DAD) was the most common histological feature in non-survivors. Furthermore, haemorrhagic necrosis and lymphocyte depletion were found in lymph nodes and the spleen, indicating a pathological basis of lymphocytopenia. However, micro thrombosis was primarily found in extrapulmonary organs in SARS-CoV-2, something which was less common in previous SARS outbreaks.
Varga et al. [12] studied the mechanism responsible for vascular dysfunction by examining the post-mortem of three COVID-19 patients. There were viral inclusion structures in the endothelial cells, which could be due to the viral targeting of angiotensinogen converting enzyme 2 (ACE2) receptors present on endothelial cells. Endothelial dysfunction occurred by the recruitment of immune system mediators, inducing damage and apoptosis. As the vascular endothelium is critical for maintaining vascular tone and homeostasis, dysfunction increases vasoconstriction, leading to ischemia and inflammatory consequences such as oedema and pro-coagulation. COVID-19 induced endotheliitis was attributed to the impairment in microcirculation occurring in various vascular beds. The risk for endothelial damage was higher with pre-existent risk factors such as smoking, hypertension, diabetes, obesity, and chronic cardiovascular illness.
A similar study by Menter et al. [13] showed signs of thrombotic microangiopathy in three out of 21 autopsy reports of COVID-19 patients. Microthrombi were found in glomerular capillaries accompanying signs of disseminated intravascular coagulation (DIC) leading to shock. They found that coagulopathies were predominantly associated with blood group A. ABO alleles may elevate the Von-Willebrand factor by 20 % and increase the risk of venous thrombosis [14,15]. The coronavirus responsible for SARS was seen to interact with blood group A antigen and viral S proteins, allowing for virus and ACE2 receptor interaction and viral entry [16]. These coagulopathies are consistent with complement-mediated microvascular damage to the lungs and skin, resulting in DAD from microthrombi and florid vasculitis of small veins and capillaries. However, vasculitis could have developed independently from bronchopneumonia as vascular changes in other organs were not found.
Lax et al. [17] examined both lungs of COVID-19 patients and found DAD and thrombosis in small and mid-sized pulmonary arteries associated with infarction and bronchopneumonia. They concluded that death might be due to thrombosis in segmental and subsegmental pulmonary arterial vessels despite prophylactic anticoagulation.
Simultaneously, several studies highlighted the effects of COVID-19 infection on platelets [4, 6, 7, 18,19,20]. Platelet count was significantly lower in non-survivors compared with survivors [18]. Guan et al. [4] reported thrombocytopenia in 36.2 % of patients on admission and 57.7 % with severe disease amongst 1099 COVID-19 patients. Another large study with 1476 patients in Wuhan, China showed a significantly higher rate of thrombocytopenia in non-survivors than survivors (72.7 % vs. 10.7 %, p<0.001) [20]. DAD by the virus could entrap megakaryocytes and hinders the release of platelets [20]. However, decreased platelet count could cause a reactive elevation in thrombopoietin levels, resulting in a hypercoagulable state [21].
Likewise, the incidence and characteristics of venous thromboembolic disease in COVID-19 patients have been the subject of numerous studies. During the initial stage of the pandemic, a COVID-19 positive patient with cerebral venous sinus thrombosis was treated successfully with low molecular weight heparin (LMWH), [22] which suggested the possibility of a thrombotic event as the initial presentation in COVID-19.
Studies have approximated 20 % risk of venous thromboembolism (VTE) and 3 % risk of stroke, especially in the severely ill and those admitted to intensive care units (ICU) 23,24,25,26,27,28]. Furthermore, COVID-19 patients were twice as likely to develop pulmonary embolism (PE) when compared to the control group. 29 While the risk of VTE appears higher in patients requiring ICU compared to those on general wards, thromboembolic events also contribute to morbidity and mortality in the ambulatory setting [30,31].
Current evidence suggests that elevated D-dimer measurements are a predictor of coagulopathy in COVID-19, [4,5,6,7,8,9] and higher D-dimer levels are directly linked to increased mortality [5,6,18]. A prospective Italian study [9] showed elevated procoagulant factors and higher D-dimer levels at baseline in all patients. Notably, a study including 1099 COVID-19 patients revealed that patients with severe infection had higher D-dimer levels [4]. A level of 0.5 mg/L or more was seen more commonly in patients with severe presentations than in those with non-severe illness (59.6 % vs. 43.2 %; P=0.002) [4]. Furthermore, D-dimer levels in ICU patients were 2.5 to 5 times higher than in non-ICU patients [6,7].
Moreover, a retrospective analysis of the coagulation function of 303 patients with COVID-19 was performed to compare the coagulation trends of the mild and severe infection [10]. The majority of patients in the severe groups were males (76.9 % vs. 49.8 %), elderly (median age: 65 vs. 50), and had underlying diseases (73.1 % vs. 36.1 %). There were abnormal coagulation parameters in 69 % of patients on admission (100 % severe group vs. 66.1 % mild group). International normalized ratio (INR), prothrombin time (PT), activated partial thromboplastin time (APTT), fibrinogen and fibrinogen degradation products, and D-dimer were higher in the severe group. Fibrinogen and APTT returned to normal as the disease improved.
However, COVID-19 patients also have an added bleeding risk [32,33]. A multicentre retrospective study [32] of 400 patients admitted to hospital with COVID-19 (144 critically ill) has shown 4.8 % overall and 2.3 % major bleeding rates. Among the critically ill patients, major bleeding rates were 5.6 % [32]. Deep muscle (23 %), gastrointestinal (14 %), central venous catheter (14 %), and otorhinolaryngological (14 %) haemorrhages have been reported [33].
Pregnant women, children and COVID 19
Pregnant women with COVID-19 are found to have increased APTT, fibrinogen, D-dimer, PT, and platelet [34]. Unfortunately, there are reports of increased stillbirth during the current COVID-19 crisis [35,36]. However, it is yet early to implicate these findings to the vascular complications without accessing the impact of disruptions in health services and reduced access to medical care during the ongoing pandemic [37].
COVID-19 positive or exposed children have displayed symptoms consistent with pediatric inflammatory conditions, known as Multisystem Inflammatory Syndrome in Children (MIS-C) [38]. This syndrome closely resembles Kawasaki disease and show elevated inflammatory markers at the time of admission with signs of coagulopathy, elevated D-dimer, and PT [38]. Most of the patients suffered from neurological and/or coagulopathic complications, requiring anticoagulation [38,39,40,41].
Mechanism of coagulopathies
SARS-CoV-2 activates coagulation, inducing procoagulant factors and proinflammatory cytokines that predispose to atherosclerotic plaque rupture, ischemia, and thrombosis [18,42]. High levels of proinflammatory cytokines/interleukins (IL) (IL2, IL7, etc.) have been observed in COVID-19 patients admitted to ICU [43]. Furthermore, coagulation abnormalities due to an inflammatory response to the virus resulting in inflammation-induced thrombosis are more apparent in those with severe disease, and those developing sepsis-induced coagulopathy and DIC [44].
The aetiology of the procoagulant response is complex and has been thought to result from specific interactions between host defense mechanisms and coagulation systems, including microangiopathy, local thrombus formation, and a systemic coagulation defect that leads to large vessel thrombosis and major venous and arterial thromboembolic complications [45,46]. Fogarty et al. [19] suggested the ‘double hit’ hypothesis for COVID-19 pulmonary manifestations, which targeted both ventilation and perfusion; the latter in the form of pulmonary vasculopathy that causes what is now known as ‘pulmonary intravascular coagulation’ or ‘PIC’.
Furthermore, Bowles et al. [47] found 31 out of 34 COVID-19 patients tested positive for lupus anticoagulant assays; dilute Russell’s viper-venom time, and lupus anticoagulant-sensitive aPTT. All patients with a positive lupus anticoagulant assay had a prolonged aPTT. However, 91 % of patients with prolonged aPTT were lupus anticoagulant positive, the majority of those having a related factor XII deficiency. The presence of lupus anticoagulant could be linked with a thrombotic tendency within the antiphospholipid syndrome. However, the role of lupus anticoagulant in the pathophysiology of COVID-19 is not yet clear.
Serological analysis has also shown the presence of anticardiolipin IgA antibodies as well as anti–β2-glycoprotein I IgA and IgG antibodies in COVID-19 patients admitted to ICU [48]. While these antibodies are central for the diagnosis of antiphospholipid syndrome, they can be positive in critically ill patients who may suffer from other causes of thrombosis, such as DIC.
Lillicrap [49] explained that the pathophysiology of DIC and the bleeding tendency in COVID-19 is complex and multifactorial, involving an interplay between cellular and plasmatic elements of the haemostatic system and components of the immune response towards the pathogen causing the infection. A dysregulated thrombin generation occurs both systemically and locally in the lungs of COVID-19 patients, resulting in the deposition of fibrin. This leads to tissue damage and microangiopathic pathology. Evolving evidence suggests that the cause of this pathophysiology is initiated by the exposure of endothelium, platelets, and leukocytes to the pathogen. This is further exacerbated by inhibition of fibrinolysis and impairment of normal anticoagulant mechanisms. However, treatment of DIC up to this publication had been primarily focused on targeting the responsible pathogen with supportive care to maintain critical organ function.
Anticoagulation therapy
Understanding the pathogenesis, epidemiology, management, and outcomes of patients with COVID-19 who develop venous or arterial thrombosis and those with a pre-existing thrombotic disease who develop COVID-19 provided a supplement for clinical-decision making in terms of antithrombotic therapy in COVID-19 patients [50].
There is a recommendation to strictly apply VTE screening, and thrombosis prophylaxis or early therapeutic anticoagulation in all COVID-19 patients admitted to ICU with severe COVID-19 infection [51,52].
There is a narrow therapeutic index between preventing and treating venous and arterial thrombotic events and the risk of bleeding in COVID-19 patients. Watson et al. [53] stated that it is reasonable to continue outpatient anticoagulation in COVID-19 confirmed or suspected patients. However, bridging should not be offered in patients with a low risk of thrombosis due to the risk of bleeding. Routine use of aspirin increases the bleeding risk and benefits should be carefully assessed. Similarly, they suggested that prolonged dual antiplatelet therapy (DAPT) beyond six months in COVID-19 after stenting and beyond a year for the acute coronary syndrome (ACS) should be weighed, balancing risks and benefits. Furthermore, warfarin-based triple therapy for COVID-19 patients with atrial fibrillation is inferior to a direct oral anticoagulant-based regimen with P2Y12 receptor blockers and short-duration aspirin [53].
Questions have been asked about a possible role for thromboprophylaxis after hospital discharge and in selected non-hospitalised COVID-19 if immobilised or they have other prothrombotic factors [54]. However, a COVID-19 positive patient has been documented to develop superior mesenteric arterial thrombosis after cessation of LMWH [55]. Khan et al. [56] suggested maintaining full anticoagulation for at least one to two months post-hospital discharge with LMWH due to its anti-inflammatory effects. Analytical parameters such as high D-dimer and IL-6 levels should also be taken into account and treatment individualised.
Recent recommendations from the International Society on Thrombosis and Hemostasis suggest that all hospitalised patients with COVID-19 should receive thromboprophylaxis or full therapeutic anticoagulation if indicated [54]. Their interim guidance on recognising and managing coagulopathy in COVID-19 called for careful reconsideration of anticoagulation, mainly focusing on the proposed approach for the use of heparin [57]. They also called for an expeditious randomised control trial of COVID-19 coagulopathy to evaluate unfractionated heparin (UH) and LMWH at therapeutic and prophylactic doses. This is with the hope of answering the critical questions surrounding anticoagulation in these patients due to the advanced form of COVID-19 showing evidence of DIC with a thrombotic phenotype. However, drug-drug interactions between antiplatelet and anticoagulant agents should be considered with investigational COVID-19 therapies [50].
Extracorporeal membrane oxygenation (ECMO)
ECMO has been tried in COVID-19 patients with respiratory failure following futile mechanical ventilation efforts. However, ECMO has been associated with thrombotic events due to activation of the coagulation cascade from non-endothelial surface contact in the extracorporeal circuit [58,59].
Extracorporeal Life Support Organization (ELSO) Registry showed significantly higher in-hospital mortality in patients who used ECMO for circulatory support (HR 1·89); however, COVID-19 patients on respiratory venovenous ECMO with concurrent ARDS had less than 40 % estimated cumulative in-hospital mortality at 90 days [60].
Higher thromboembolic risk in patients on ECMO favours the routine anticoagulation use [59, 61]. ELSO COVID-19 Interim Guidelines support implementing the standard ELSO anticoagulation guidelines in COVID-19 patients [62]. Systemic anticoagulation with bivalirudin on venovenous ECMO had lower circuit-related thrombotic events [63]. However, Parzy et al. [64] reported that all their patients with SARS-CoV2 infection and ARDS, who were on venovenous ECMO, suffered from venous thromboembolism despite receiving the anticoagulation.
Anticoagulation strategy in COVID-19 patients on ECMO is further complicated by the co-existence of thrombocytopenia and thromboembolism [65,66]. Therefore, the innate thrombotic potential of ECMO within the emerging evidence of higher thrombotic complications seen in COVID-19 makes it challenging, necessitating a large scale study to establish its clinical utility [67,68].
Risk stratification
There is a need to determine the role of biomarkers and/or scoring systems to stratify patients’ risk in COVID-19 [23]. Older age, high ‘Sequential Organ Failure Assessment’ (SOFA) score, and D-dimer greater than one ug/mL were associated with poor prognosis [18]. Patients who died were more likely to be elderly, have more pre-existing morbidities, dyspnoea, low oxygen saturation, increased white blood cell count, decreased lymphocytes, and elevated C-reactive protein (CRP) levels [69]. Furthermore, complications such as ARDS, acute cardiac injury, acute kidney injury, shock, and DIC were more commonly seen in non-survivors [69].
Fibrinolysis shutdown evident by elevated D-dimer levels and complete failure of clot lysis at 30 min on thromboelastography predicts thromboembolic events and the need for haemodialysis in critically ill patients with COVID-19 [70].
Khan et al. [56] suggested that D-dimer levels and renal function should guide the anticoagulation in the ICU. For patients with D-dimer levels >1000ug/mL, enoxaparin 1.5 mg/kg once daily was initiated. If the patient’s creatinine clearance was <30mL/min, 1 mg/kg was used until the D-dimer level was <1000ug/mL. However, this treatment was continued if there was clinical evidence of deep venous thrombosis (DVT), PE, or other thrombotic events regardless of D-dimer level. D-dimer levels were evaluated daily to assess the VTE risk, and clinical examinations were performed to look for any signs. They reported no instances of PE and one case of DVT.
Although D-dimer is a valuable tool in categorizing the risk of developing VTE in patients with novel coronavirus pneumonia (NCP), a higher threshold D-dimer is suggested for the detection of PE [28,71]. In the presence of clinical signs and/or suspicion of VTE, compression ultrasonography and echocardiography should be performed irrespective of disease stage [72]. Furthermore, patients with severe COVID-19 may have associated PE, and thus, the use of contrast computerized tomography (CT) rather than routine non-contrast CT is encouraged [73].
Discussion
COVID-19 can lead to severe illness with accompanying multiorgan failure. Inflammatory cytokine release mediating atherosclerosis results in hemodynamic changes and causes induction of procoagulant mediators. It is believed that a storm of inflammatory mediators triggered by SARS-CoV-2 results in irreversible multisystem inflammation that can partially present as abnormal haemostasis and coagulopathy, further aggravating organ injury [74]. Similarly, the virus targets ACE-2 receptors present on vascular muscle membrane and endothelial cells [75].
The pathogenesis of thrombotic events induced by COVID-19 could occur due to host cell aggression [47,76]. An excessive immune-response induced cytokine storm is followed by inflammatory response activation and hypercoagulability, ultimately resulting in systemic thrombosis formation. Complications could include DIC, although not common. Similarly, pulmonary micro-thrombosis can induce COVID-19 related ARDS. Monitoring critically ill patients includes evaluating PT, fibrinogen, platelet, and D-dimer levels. Elevated D-dimer and fibrinogen levels suggest a hypercoagulable and inflammatory state. High D-dimer levels are also a predictor of ARDS, which leads to increased mortality.
Blood clots can, to some degree, act as a barrier to pathogen invasion, referred to as immunothrombosis. However, this immunothrombosis concept is affected by COVID-19 [77]. Haemostatic components initially operate in a chemotactic fashion, which stimulates the immune system. As SARS-CoV-2 targets, the lungs, bronchoalveolar haemostasis activation attempts to combat the infection by recruiting immune cells [77]. This can be observed by elevated acute phase reactants such as platelets, fibrinogen, and CRP. When examining post-mortem reports, the presence of pulmonary microthrombi also points to hypercoagulability induced by SARS-CoV-2. Among those patients with mild-moderate disease, the microthrombi are broken down, presenting as an elevation in D-dimer levels. In severe disease, marked activation of the coagulation system could lead to thrombosis in limbs, intestines, and the brain.
Unfortunately, those vulnerable to thrombotic events are more likely to have severe COVID-19 illness [77]. Elevations in platelet count and acute phase reactants, such as fibrinogen, lead to hypercoagulability in these patients. Microthrombi formation, as a result of this hypercoagulable state, is linked to patient deterioration. In mild illness, microthrombi are broken down by the fibrinolytic function of the lungs. Alternatively, in severe infection, the pulmonary coagulation system is highly activated, leading to possible systemic thrombosis in limbs, intestine, and brain with resultant multiorgan damage.
Complement-mediated thrombotic microangiopathy (TMA) in severe infection is possible [78]. TMA is characterised by microangiopathic haemolytic anaemia, thrombocytopenia, and organ damage such as neurological, renal, and cardiac dysfunction. However, reports of schistocytes to confirm microangiopathic haemolytic anaemia is lacking, and little is known about the nature of cardiac dysfunction in COVID-19 patients. As such, the pathophysiology of TMA is described in a two-hit hypothesis. The first hit is a germline mutation in complement regulatory protein, and the second hit could be due to pregnancy, inflammation, surgery, or autoimmunity.
Diurno et al. [79] treated four severe COVID-19 patients with eculizumab (one of the two FDA-approved complement inhibitors: eculizumab in 2007 and ravulizumab in 2019) resulting in immediate improvement and successful disease outcomes. This highlights a need for more extensive prospective trials to examine complement inhibitors in patients with severe COVID-19 infection. Furthermore, the concern of thromboembolic risk and microvascular thrombosis in patients with COVID-19 suggests a need for RCT to examine the possible benefit of prophylactic anticoagulation or using increased anticoagulation doses in inpatients or long-term anticoagulation following discharge [80].
Conclusions
Current evidence indicates dysfunctional coagulation pathways in COVID-19 patients; however, specific studies concerning vascular complications are lacking. It is crucial to have large-scale multicentre studies to establish and ascertain the vascular complications in COVID-19 to formulate management strategies with optimal patient outcomes.
References
Tu H, Tu S, Gao S et al (2020) Current epidemiological and clinical features of COVID-19; a global perspective from China. J Infect 81:1–9
Liu PP, Blet A, Smyth D et al (2020) The science underlying COVID-19: implications for the cardiovascular system. Circulation 142:68–78
Oudkerk M, Büller HR, Kuijpers D et al (2020) Diagnosis, prevention, and treatment of thromboembolic complications in COVID-19: report of the National Institute for Public Health of the Netherlands. Radiology 297:E216–E222
Guan WJ, Ni ZY, Hu Y et al (2020) China medical treatment expert group for Covid-19. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382:1708–1720
Tang N, Li D, Wang X et al (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 18:844–847
Wang D, Hu B, Hu C et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected Pneumonia in Wuhan, China [published correction appears in JAMA. 2021 Mar 16;325(11):1113]. JAMA 2020;323:1061-1069
Huang C, Wang Y, Li X et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China [published correction appears in Lancet. 2020 Jan 30;:]. Lancet 2020;395:497-506
Zhang Y, Cao W, Xiao M et al (2020) [Clinical and coagulation characteristics of 7 patients with critical COVID-2019 pneumonia and acro-ischemia]. Zhonghua Xue Ye Xue Za Zhi 41:E006
Ranucci M, Ballotta A, Di Dedda U et al (2020) The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost 18:1747–1751
Zou Y, Guo H, Zhang Y et al (2020) Analysis of coagulation parameters in patients with COVID-19 in Shanghai, China. Biosci Trends 14:285–289
Zhang T, Sun LX, Feng RE (2020) [Comparison of clinical and pathological features between severe acute respiratory syndrome and coronavirus disease 2019]. Zhonghua Jie He He Hu Xi Za Zhi 43:496–502
Varga Z, Flammer AJ, Steiger P et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395:1417–1418
Menter T, Haslbauer JD, Nienhold R et al (2020) Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 77:198–209
Jenkins PV, O’Donnell JS (2006) ABO blood group determines plasma von Willebrand factor levels: a biologic function after all? Transfusion 46:1836–1844
Franchini M, Mannucci PM (2014) ABO blood group and thrombotic vascular disease. Thromb Haemost 112:1103–1109
Guillon P, Clement M, Sebille V et al (2008) Inhibition of the interaction between the SARS-CoV spike protein and its cellular receptor by anti‐histo‐blood group antibodies. Glycobiology 18:1085–1093
Lax SF, Skok K, Zechner P et al (2020) Pulmonary arterial thrombosis in COVID-19 with fatal outcome: results from a prospective, single-center, clinicopathologic case series. Ann Intern Med 173:350–361
Zhou F, Yu T, Du R et al (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395:1054–1062
Fogarty H, Townsend L, Ni Cheallaigh C et al (2020) COVID19 coagulopathy in Caucasian patients. Br J Haematol 189:1044–1049
Yang X, Yang Q, Wang Y et al (2020) Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost 18:1469–1472
Yin S, Huang M, Li D et al (2021) Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J Thromb Thrombolysis 51:1107–1110
Hughes C, Nichols T, Pike M et al (2020) Cerebral venous sinus thrombosis as a presentation of COVID-19. Eur J Case Rep Intern Med 7:001691
Al-Ani F, Chehade S, Lazo-Langner A (2020) Thrombosis risk associated with COVID-19 infection. A scoping review. Thromb Res 192:152–160
Rech TH, Girardi AM, Gazzana MB (2020) Incidence of pulmonary embolism in patients with COVID-19. Intensive Care Med 46:1500–1501
Helms J, Tacquard C, Severac F et al, CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis) (2020) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 46:1089–1098
Middeldorp S, Coppens M, van Haaps TF et al (2020) Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost 18:1995–2002
Hippensteel JA, Burnham EL, Jolley SE (2020) Prevalence of venous thromboembolism in critically ill patients with COVID-19. Br J Haematol 190:e134–e137
Léonard-Lorant I, Delabranche X, Séverac F et al (2020) Acute pulmonary embolism in patients with COVID-19 at CT angiography and relationship to d-Dimer levels. Radiology 296:E189–E191
Poissy J, Goutay J, Caplan M et al (2020) Lille ICU haemostasis COVID-19 group. Pulmonary embolism in patients with COVID-19: awareness of an increased prevalence. Circulation 142:184–186
Langer F, Kluge S, Klamroth R et al (2020) Coagulopathy in COVID-19 and its implication for safe and efficacious thromboprophylaxis. Hamostaseologie 40:264–269
Oxley TJ, Mocco J, Majidi S et al (2020) Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med 382:e60
Al-Samkari H, Karp Leaf RS, Dzik WH et al (2020) COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 136:489–500
Fraissé M, Logre E, Pajot O et al (2020) Thrombotic and hemorrhagic events in critically ill COVID-19 patients: a French monocenter retrospective study. Crit Care 24:275
Koumoutsea EV, Vivanti AJ, Shehata N et al (2020) COVID-19 and acute coagulopathy in pregnancy. J Thromb Haemost 18:1648–1652
Watson C (2020) Stillbirth rate rises dramatically during pandemic. Nature 585:490–491
Khalil A, von Dadelszen P, Draycott T et al (2020) Change in the incidence of stillbirth and preterm delivery during the COVID-19 pandemic. JAMA 324:705–706
Homer CSE, Leisher SH, Aggarwal N et al (2021) Counting stillbirths and COVID 19-there has never been a more urgent time. Lancet Glob Health 9:e10–e11
Lee PY, Day-Lewis M, Henderson LA et al (2020) Distinct clinical and immunological features of SARS-CoV-2-induced multisystem inflammatory syndrome in children. J Clin Invest 130:5942–5950
Dufort EM, Koumans EH, Chow EJ et al (2020) New York State and Centers for Disease Control and Prevention Multisystem Inflammatory Syndrome in Children Investigation Team. Multisystem Inflammatory Syndrome in Children in New York State. N Engl J Med 383:347–358
Bhatta S, Sayed A, Ranabhat B et al. New-Onset Seizure as the Only Presentation in a Child With COVID-19. Cureus 2020;12:e8820
Moraleda C, Serna-Pascual M, Soriano-Arandes A, EPICO-AEP Working Group et al (2021) Multi-inflammatory syndrome in children related to severe acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in Spain. Clin Infect Dis 72:e397–e401
Sultan S, Sultan M (2020) COVID-19 cytokine storm and novel truth. Med Hypotheses 144:109875
Harenberg J, Favaloro E (2020) COVID-19: progression of disease and intravascular coagulation - present status and future perspectives. Clin Chem Lab Med 58:1029–1036
Connors JM, Levy JH (2020) COVID-19 and its implications for thrombosis and anticoagulation. Blood 135:2033–2040
Di Micco P, Russo V, Carannante N et al (2020) Clotting factors in COVID-19: epidemiological association and prognostic values in different clinical presentations in an Italian Cohort. J Clin Med 9:1371
Iba T, Levy JH, Levi M et al (2020) Coagulopathy of coronavirus disease 2019. Crit Care Med 48:1358–1364
Bowles L, Platton S, Yartey N et al (2020) Lupus anticoagulant and abnormal coagulation tests in patients with Covid-19. N Engl J Med 383:288–290
Zhang Y, Xiao M, Zhang S et al (2020) Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med 382:e38
Lillicrap D (2020) Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J Thromb Haemost 18:786–787
Bikdeli B, Madhavan MV, Jimenez D et al; Global COVID-19 Thrombosis Collaborative Group, Endorsed by the ISTH, NATF, ESVM, and the IUA, Supported by the ESC Working Group on Pulmonary Circulation and Right Ventricular Function. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J Am Coll Cardiol 2020;75:2950-2973
Klok FA, Kruip MJHA, van der Meer NJM et al (2020) Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb Res 191:148–150
Llitjos JF, Leclerc M, Chochois C et al (2020) High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost 18:1743–1746
Watson RA, Johnson DM, Dharia RN et al (2020) Anti-coagulant and anti-platelet therapy in the COVID-19 patient: a best practices quality initiative across a large health system. Hosp Pract (1995) 48:169–179
Kollias A, Kyriakoulis KG, Dimakakos E et al (2020) Thromboembolic risk and anticoagulant therapy in COVID-19 patients: emerging evidence and call for action. Br J Haematol 189:846–847
Beccara A, Pacioni L, Ponton C, Francavilla S, Cuzzoli S (2020) Arterial mesenteric thrombosis as a complication of SARS-CoV-2 infection. Eur J Case Rep Intern Med 7:001690
Khan IH, Savarimuthu S, Leung MST et al (2020) The need to manage the risk of thromboembolism in COVID-19 patients. J Vasc Surg 72:799–804
Barrett CD, Moore HB, Yaffe MB et al (2020) ISTH interim guidance on recognition and management of coagulopathy in COVID-19: A comment. J Thromb Haemost 18:2060–2063
Murphy DA, Hockings LE, Andrews RK et al (2015) Extracorporeal membrane oxygenation-hemostatic complications. Transfus Med Rev 29:90–101
Beyls C, Huette P, Abou-Arab O et al (2020) Extracorporeal membrane oxygenation for COVID-19-associated severe acute respiratory distress syndrome and risk of thrombosis. Br J Anaesth 8:1121–1131
Barbaro RP, MacLaren G, Boonstra PS et al. Extracorporeal membrane oxygenation support in COVID-19: an international cohort study of the Extracorporeal Life Support Organization registry [published correction appears in Lancet. 2020 Oct 10;396(10257):1070]. Lancet 2020;396:1071-1078
Guo Z, Sun L, Li B et al (2021) Anticoagulation management in severe coronavirus disease 2019 patients on extracorporeal membrane oxygenation. J Cardiothorac Vasc Anesth 35:389–397
Shekar K, Badulak J, Peek G et al (2020) Extracorporeal life support organization coronavirus disease 2019 interim guidelines: a consensus document from an International group of interdisciplinary extracorporeal membrane oxygenation providers. ASAIO J 66:707–721
Rivosecchi RM, Arakelians AR, Ryan J et al (2021) Comparison of Anticoagulation Strategies in Patients Requiring Venovenous Extracorporeal Membrane Oxygenation: Heparin Versus Bivalirudin. Crit Care Med 49:1129–1136
Parzy G, Daviet F, Puech B et al (2020) Venous Thromboembolism Events Following Venovenous Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Syndrome Coronavirus 2 Based on CT Scans. Crit Care Med 48:e971–e975
Jones C, Chen K, Narendran VC, Series (2021) The Coexistence of Thrombocytopenia and Thromboembolism in COVID-19 Patients on ECMO: A Case Series and Literature Review. Front Med (Lausanne) 8:731352
Steinlechner B, Kargl G, Schlömmer C et al (2021) Can Heparin-Coated ECMO Cannulas Induce Thrombocytopenia in COVID-19 Patients? Case Reports Immunol 2021:6624682
Doyle AJ, Hunt BJ, Sanderson B et al (2021) A Comparison of Thrombosis and Hemorrhage Rates in Patients With Severe Respiratory Failure Due to Coronavirus Disease 2019 and Influenza Requiring Extracorporeal Membrane Oxygenation. Crit Care Med 49:e663–e672
Zhang Y, Ji B, Zhou Z (2021) ECMO support for COVID-19: a balancing act. Lancet 397:94–95
Deng Y, Liu W, Liu K et al (2020) Clinical characteristics of fatal and recovered cases of coronavirus disease 2019 in Wuhan, China: a retrospective study. Chin Med J (Engl) 133:1261–1267
Wright FL, Vogler TO, Moore EE et al (2020) Fibrinolysis shutdown correlation with thromboembolic events in severe COVID-19 infection. J Am Coll Surg 231:193-203.e1
Cui S, Chen S, Li X et al (2020) Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost 18:1421–1424
Poggiali E, Bastoni D, Ioannilli E et al (2020) Deep vein thrombosis and pulmonary embolism: two complications of COVID-19 Pneumonia? Eur J Case Rep Intern Med 7:001646
Grillet F, Behr J, Calame P et al (2020) Acute pulmonary embolism associated with COVID-19 Pneumonia detected with pulmonary CT angiography. Radiology 296:E186–E188
Perini P, Nabulsi B, Massoni CB et al (2020) Acute limb ischaemia in two young, non-atherosclerotic patients with COVID-19. Lancet 395:1546
Liu J, Li S, Liu J et al (2020) Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 55:102763
Joly BS, Siguret V, Veyradier A (2020) Understanding pathophysiology of hemostasis disorders in critically ill patients with COVID-19. Intensive Care Med 46:1603–1606
Thachil J, Agarwal S (2020) Understanding the COVID-19 coagulopathy spectrum. Anaesthesia 75:1432–1436
Gavriilaki E, Brodsky RA, Severe (2020) COVID-19 infection and thrombotic microangiopathy: success does not come easily. Br J Haematol 189:e227–e230
Diurno F, Numis FG, Porta G (2020) al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci 24:4040–4047
Thomas W, Varley J, Johnston A et al (2020) Thrombotic complications of patients admitted to intensive care with COVID-19 at a teaching hospital in the United Kingdom. Thromb Res 191:76–77
Acknowledgements
None.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sector.
Author information
Authors and Affiliations
Contributions
All the authors were involved in the study conception and design, acquisition, analysis and interpretation of studies and review, drafting of article, and the critical revision. All the authors approve the final submission of this article.
Corresponding author
Ethics declarations
Conflict of interest
The Authors declare that there is no conflict of interest.
Ethical approval
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Acharya, Y., Alameer, A., Calpin, G. et al. A comprehensive review of vascular complications in COVID-19. J Thromb Thrombolysis 53, 586–593 (2022). https://doi.org/10.1007/s11239-021-02593-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-021-02593-2