
The Spike Protein of SARS-CoV-2 Is Adapting Because of Selective Pressures

 ,
,  , and
, and
Abstract
:1. Introduction
2. Data Sources
3. Materials and Methods
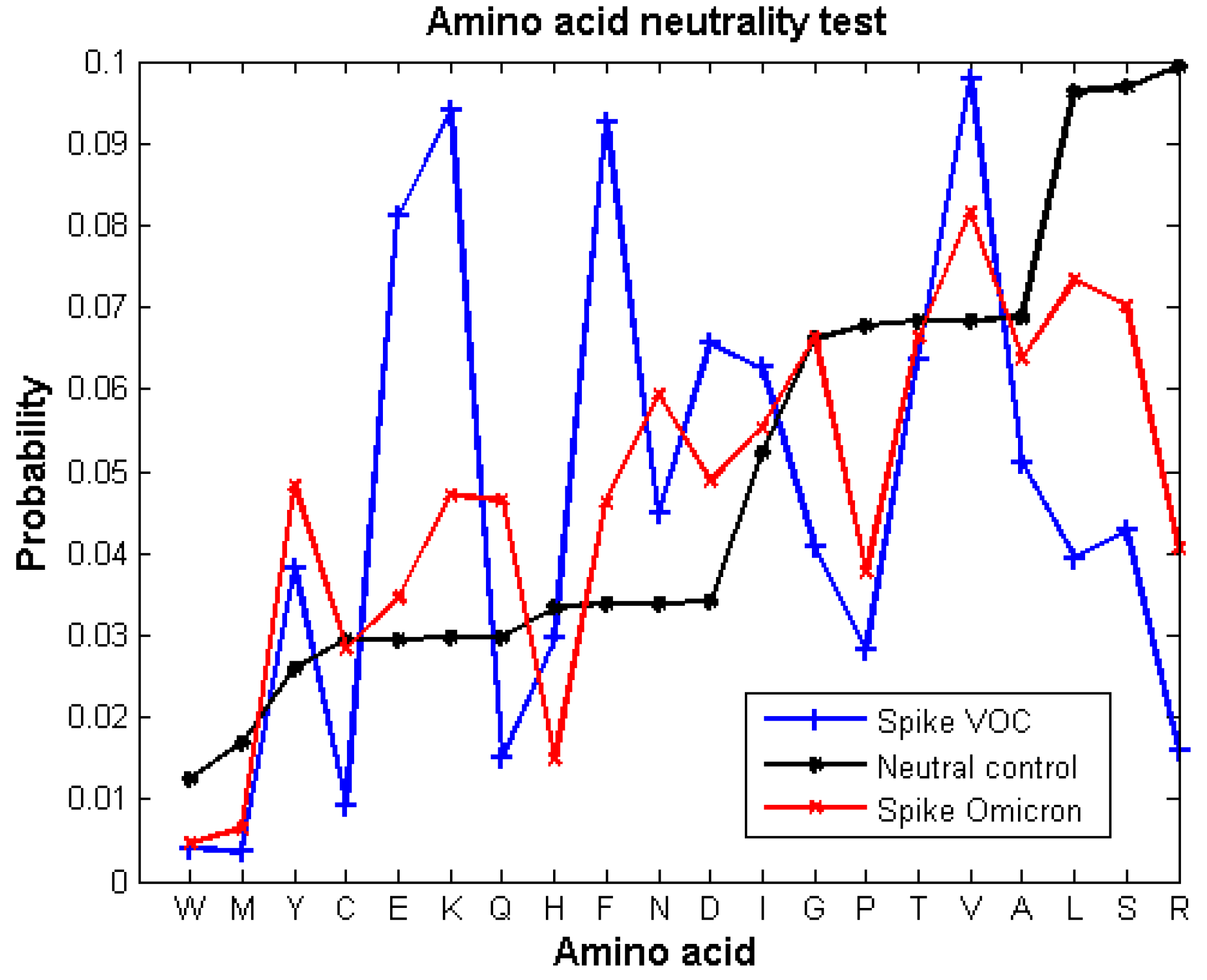
3.1. Neutral Evolution Test
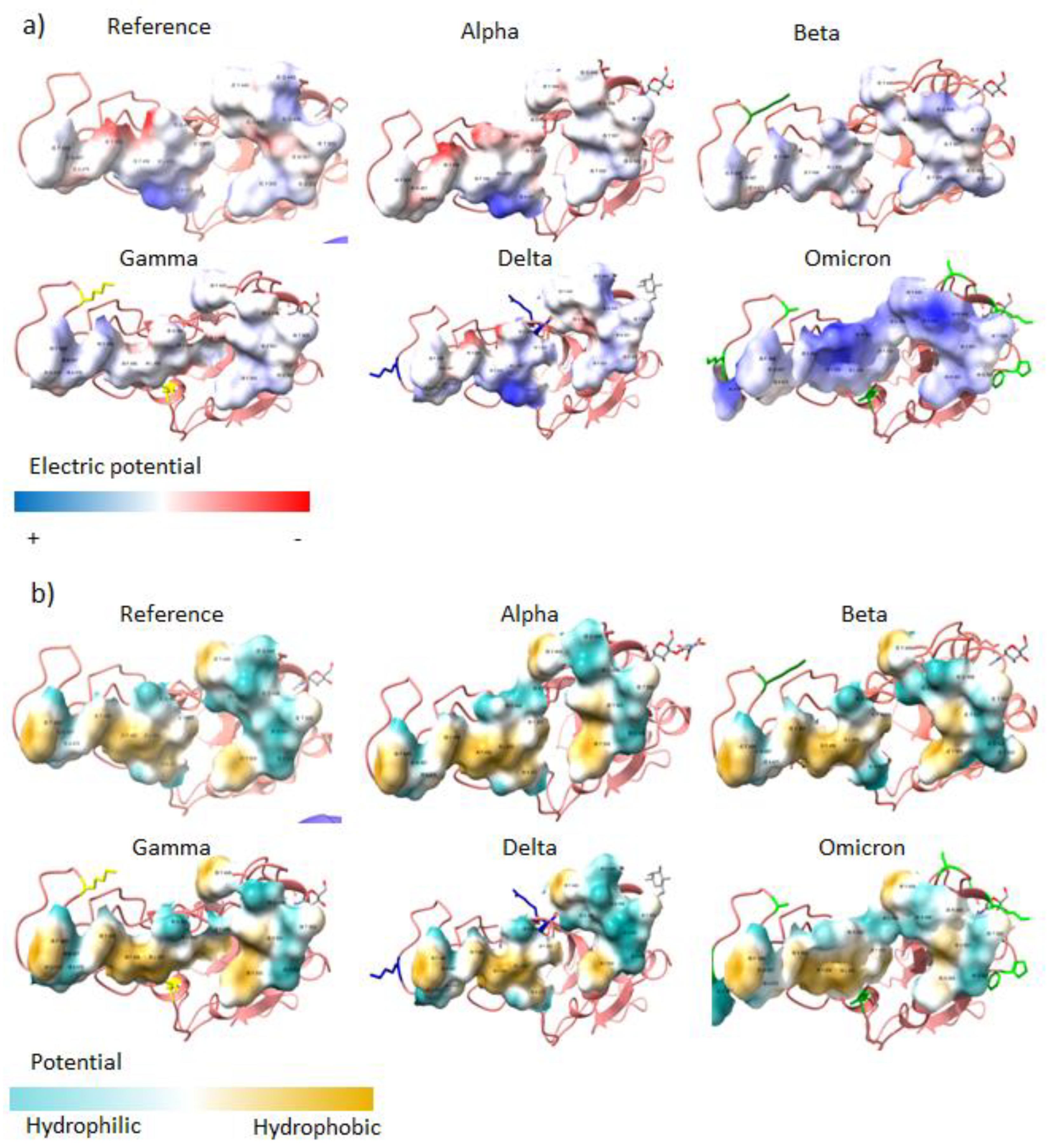
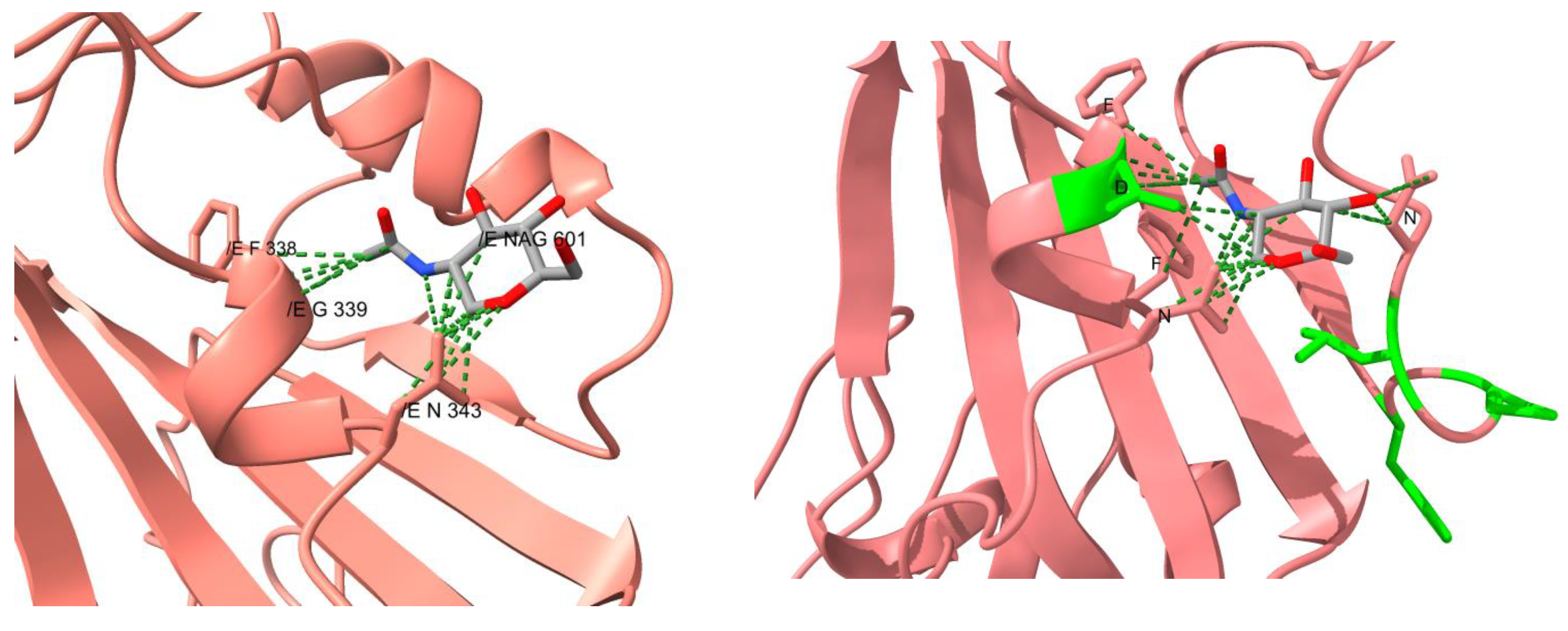
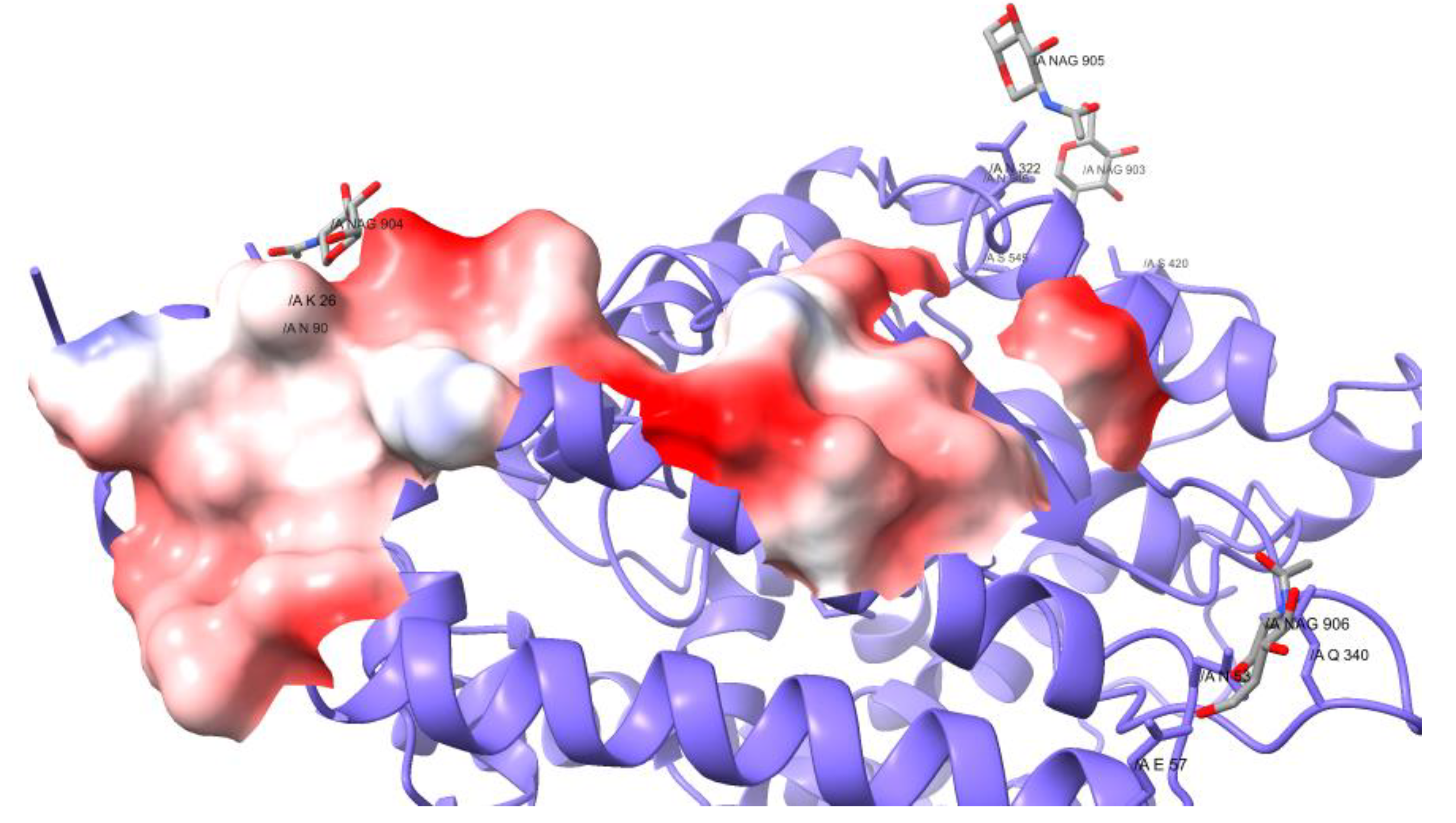
3.2. Structural Analysis
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khandia, R.; Singhal, S.; Alqahtani, T.; Kamal, M.A.; El-Shall, M.A.; Nainu, F.; Desingu, P.A.; Dhama, K. Emergence of SARS-CoV-2 Omicron (B.1.1.529) variant, salient features, high global health concerns and strategies to counter it amid ongoing COVID-19 pandemic. Environ. Res. 2022, 209, 112816. [Google Scholar] [CrossRef]
- Elliott, P.; Bodinier, B.; Eales, O.; Wang, H.W.; Haw, D.; Elliott, J.; Whitaker, M.; Jonnerby, J.; Tang, D.; Walters, C.E.; et al. Rapid increase in Omicron infections in England during December 2021: REACT-1 study. Science 2022, 375, 1406–1411. [Google Scholar] [CrossRef]
- Dhawan, M.; Choudhary, P.; Choudhary, O.P. Emergence of Omicron sub-variant BA.2: Is it a matter of concern amid the COVID-19 pandemic? Int. J. Surg. 2022, 99, 106581. [Google Scholar] [CrossRef]
- Pulliam, J.; Schalkwyk, C.; Govender, N.; Gottberg, A.V.; Cohen, C.; Groome, M.J.; Dushoff, J.; Mlisana, K.; Moultrie, H. Increased risk of SARS-CoV-2 reinfection associated with emergence of Omicron in South Africa. Science 2022, 376, 4947. [Google Scholar] [CrossRef]
- Bolze, A.; Cirulli, E.T.; Luo, S.S.; White, S.; Wyman, D.; Rossi, A.D.; Machado, H.; Cassens, T.; Jacobs, S.; Barrett, K.M.S.; et al. Rapid displacement of SARS-CoV-2 variant B.1.1.7 by B.1.617.2 and P.1 in the United States. medRxiv 2021. [Google Scholar] [CrossRef]
- Desingu, P.A.; Nagarajan, K.; Dhama, K. Emergence of Omicron third lineage BA.3 and its importance. J. Med. Virol. 2022, 94, 1808–1810. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, G.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.L.; Oliveira, T.; Pond, S.L.K.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Y.Y.; Xu, P.Y.; Cao, X.D.; Wu, C.R.; Gu, C.Y.; He, X.H.; Wang, X.X.; Huang, S.J.; Yuan, Q.N.; et al. Structures of the Omicron Spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef]
- Liu, J.; Chandrashekar, A.; Sellers, D.; Barrett, J.; Jacob-Dolan, C.; Lifton, M.; McMahan, K.; Sciacca, M.; VanWyk, H.; Wu, C.; et al. Vaccines elicit highly conserved cellular immunity to SARS-CoV-2 Omicron. Nature 2022, 603, 493–496. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.J.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of Antibody Immunity to SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef]
- Vázquez-Jiménez, A.; León, U.E.A.P.E.; Matadamas-Guzman, M.; Muciño-Olmos, E.A.; Martínez-López, Y.E.; Escobedo-Tapia, T.; Resendis-Antonio, O. On Deep Landscape Exploration of COVID-19 Patients Cells and Severity Markers. Front. Immunol. 2021, 12, 3676. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2021, 602, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Zamudio, G.S.; Prosdocimi, F.; De Farias, S.T.; José, M.V. A neutral evolution test derived from a theoretical amino acid substitution model. J. Theor. Biol. 2019, 467, 31–38. [Google Scholar] [CrossRef]
- López-Cortés, G.I.; Palacios-Pérez, M.; Zamudio, G.S.; Veledíaz, H.F.; Ortega, E.; José, M.V. Neutral evolution test of the spike protein of SARS-CoV-2 and its implications in the binding to ACE2. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- José, M.V.; Zamudio, G.S.; Morgado, E.R. A unified model of the standard genetic code. R. Soc. Open Sci. 2017, 4, 160908. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic. Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Wang, H.; Huynh, T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: Insights from molecular dynamics simulations. FEBS Lett. 2021, 595, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.Y.; Plante, K.S.; Plante, J.A.; Xie, X.P.; Zhang, X.W.; Ku, Z.Q.; An, Z.Q.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 transmission. bioRxiv Prepr. Serv. Biol. 2021. [Google Scholar] [CrossRef]
- Gu, H.J.; Chen, Q.; Yang, G.; He, L.; Fan, H.; Deng, Y.Q.; Wang, Y.X.; Teng, Y.; Zhao, Z.P.; Cui, Y.J.; et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 2020, 369, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.T.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2021, 370, 1464–1468. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.Y.; Xia, H.J.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.W.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Portmann, J.; Halwe, N.J.; et al. SARS-CoV-2 spike D614G variant confers enhanced replication and transmissibility. bioRxiv 2020, 27. [Google Scholar] [CrossRef]
- Sikora, M.; von Bülow, S.; Blanc, F.E.C.; Gecht, M.; Covino, R.; Hummer, G. Computational epitope map of SARS-CoV-2 spike protein. PLoS Comput. Biol. 2021, 17, e1008790. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, H.; Wang, H. Glycans of SARS-CoV-2 Spike Protein in Virus Infection and Antibody Production. Front. Mol. Biosci. 2021, 8, 53. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Gobeil, S.M.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D.P.; et al. Structural diversity of the SARS-CoV-2 Omicron spike. Mol. Cell 2022. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.G.; Benton, D.J.; Xu, P.Q.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Saunders, K.; Edwards, R.J.; et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation and antigenicity. bioRxiv Prepr. Serv. Biol. 2021, 6226, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.L.; Clark, T.; Raman, R.; Sasisekharan, R. Insights on the mutational landscape of the SARS-CoV-2 Omicron variant receptor-binding domain. Cell Rep. Med. 2022, 3, 100527. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.G.; Amin, A.B.; Ali, A.R.; Hoots, B.; Cadwell, B.L.; Arora, S.; Avoundjian, T.; Awofeso, A.O.; Barnes, J.; Bayoumi, N.S.; et al. COVID-19 Incidence and Death Rates Among Unvaccinated and Fully Vaccinated Adults with and Without Booster Doses During Periods of Delta and Omicron Variant Emergence-25 U.S. Jurisdictions, April 4–December 25, 2021. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Risk, M.; Schiopu, R.; Hayek, S.S.; Xie, T.K.; Holevinski, L.; Akin, C.; Freed, G.; Zhao, L.L. Efficacy of COVID-19 vaccines in patients taking immunosuppressants. Ann. Rheum. Dis. 2020, 81, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Nemet, I.; Kliker, L.; Lustig, Y.; Zuckerman, N.; Erster, O.; Cohen, C.; Kreiss, Y.; Alroy-Preis, S.; Regev-Yochay, G.; Mendelson, E.; et al. Third BNT162b2 Vaccination Neutralization of SARS-CoV-2 Omicron Infection. N. Engl. J. Med. 2022, 386, 492–494. [Google Scholar] [CrossRef]
- Wang, C.Y.; Huang, K.P.; Kuo, H.K.; Peng, W.J.; Shen, Y.H.; Kuo, B.S.; Huang, J.H.; Liu, H.; Ho, Y.H.; Lin, F.; et al. A multitope SARS-COV-2 vaccine provides long-lasting B cell and T cell immunity against Delta and Omicron variants. J. Clin. Investig. 2022, 132, e157707. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Denis, K.J.S.; Hoelzemer, A.; Lam, E.C.; Nitido, A.D.; Sheehan, M.L.; Berrios, C.; Ofoman, O.; Chang, C.C.; Hauser, B.M.; et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 2022, 185, 457–466.e4. [Google Scholar] [CrossRef]
- Flemming, A. Cross reactive T cells hold up against Omicron. Nat. Rev. Immunol. 2022, 22, 146. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.P.Y.; Ho, J.C.W.; Cheung, M.C.; Ng, K.C.; Ching, R.H.H.; Lai, K.L.; Kam, T.T.; Gu, H.G.; Sit, K.Y.; Hsin, M.K.Y.; et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 2022, 603, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, Y.F.; Lavine, C.L.; Peng, H.Q.; Zhu, H.S.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; Rits-Volloch, S.; et al. Structural and functional impact by SARS-CoV-2 Omicron spike mutations. Cell Rep. 2022, 39, 110729. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.S.; Lavine, C.L.; Rawson, S.; Peng, H.Q.; Zhu, H.S.; Anand, K.; Tong, P.; Gautam, A.; et al. Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. bioRxiv Prepr. Serv. Biol. 2021, 9745, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Mohamed, M.E.M.; El-Lateef, H.M.A.; Venugopala, K.N.; El-Beltagi, H.S. Omicron variant genome evolution and phylogenetics. J. Med. Virol. 2022, 94, 1627–1632. [Google Scholar] [CrossRef]
- Sarkar, R.; Lo, M.; Saha, R.; Dutta, S.; Chawla-Sarkar, M. S glycoprotein diversity of the Omicron variant. medRxiv 2021. Available online: http://medrxiv.org/content/early/2021/12/06/2021.12.04.21267284.abstract (accessed on 20 May 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Alpha | Beta | Gamma | Delta | Omicron | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 339 | G | D | + | ||||||||
| 371 | S | L | - | ||||||||
| 373 | S | P | - | ||||||||
| 375 | S | F | + | ||||||||
| 417 | K | N | + | T | + | N | + | ||||
| 440 | N | K | + | ||||||||
| 446 | G | S | - | ||||||||
| 452 | L | R | - | ||||||||
| 477 | S | N | + | ||||||||
| 478 | T | K | + | K | + | ||||||
| 484 | E | K | + | K | + | A | / | ||||
| 490 | F | ||||||||||
| 493 | Q | R | - | ||||||||
| 496 | G | S | - | ||||||||
| 498 | Q | R | - | ||||||||
| 501 | N | Y | + | Y | + | Y | + | Y | + | ||
| 505 | T | Y | + | Y | + | Y | + | H | - | ||
| 547 | T | K | + | ||||||||
| 570 | A | D | + | ||||||||
| 614 | D | G | - | G | - | G | - | G | - | G | - |
| 655 | H | Y | + | Y | + | ||||||
| 677 | Q | ||||||||||
| 679 | N | K | + | ||||||||
| 681 | P | H | - | R | - | H | - | ||||
| Reference | Alpha | Beta | Gamma | Delta | Omicron | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 417 | K | 4 | K | 1 | K | 3 | ||||||
| 446 | G | 1 | ||||||||||
| 449 | Y | 6 | Y | 5 | Y | 2 | ||||||
| 453 | Y | 1 | Y | 3 | Y | 1 | Y | 3 | Y | 3 | ||
| 455 | L | 2 | L | 3 | L | 4 | L | 2 | ||||
| 456 | F | 5 | F | 4 | F | 8 | F | 5 | F | 2 | F | 3 |
| 473 | Y | Y | 1 | Y | 1 | |||||||
| 475 | A | 2 | A | 2 | A | 1 | A | 1 | A | 1 | A | 2 |
| 476 | G | G | 1 | G | 1 | |||||||
| 486 | F | 9 | F | 5 | F | 8 | F | 6 | F | 5 | F | 7 |
| 487 | N | 3 | N | 7 | N | 5 | N | 2 | N | 6 | N | 4 |
| 489 | Y | 4 | Y | 4 | Y | 5 | Y | 5 | Y | 4 | Y | 6 |
| 493 | Q | 2 | Q | 13 | Q | 6 | Q | 2 | Q | 3 | R | 7 |
| 494 | S | S | 2 | S | 2 | S | 2 | |||||
| 496 | G | 1 | G | 2 | S | 2 | ||||||
| 498 | Q | 7 | Q | 3 | Q | 3 | Q | 4 | Q | 2 | R | 4 |
| 500 | T | 9 | T | 8 | T | 8 | T | 10 | T | 7 | T | 6 |
| 501 | N | 1 | Y | 12 | N | 5 | Y | 14 | N | 9 | Y | 14 |
| 502 | G | 5 | G | 5 | G | 1 | G | 2 | G | 4 | G | 6 |
| 503 | V | V | 1 | |||||||||
| 505 | Y | 12 | Y | 10 | T | 13 | Y | 16 | Y | 20 | H | 13 |
| SARS-CoV-2 | Alpha | Beta | Gamma | Delta | Omicron | |
|---|---|---|---|---|---|---|
| Hydrogen bonds | 10 | 8 | 2 | 6 | 12 | 13 |
| Hydrophobic contacts | 18 | 11 | 21 | 16 | 10 | 12 |
| Contacts | 74 | 77 | 72 | 72 | 80 | 82 |
| Mean distance (Å) | 3.563 | 3.572 | 3.414 | 3.569 | 3.492 | 3.557 |
| Number of residues | 17 | 14 | 16 | 13 | 17 | 16 |
| RMSD (Ǻ) | TM Score | |
|---|---|---|
| Alpha | 0.55 | 0.99 |
| Beta | 0.74 | 0.98 |
| Gamma | 0.54 | 0.99 |
| Delta | 0.55 | 0.99 |
| Omicron | 1.11 | 0.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Cortés, G.I.; Palacios-Pérez, M.; Veledíaz, H.F.; Hernández-Aguilar, M.; López-Hernández, G.R.; Zamudio, G.S.; José, M.V. The Spike Protein of SARS-CoV-2 Is Adapting Because of Selective Pressures. Vaccines 2022, 10, 864. https://doi.org/10.3390/vaccines10060864
López-Cortés GI, Palacios-Pérez M, Veledíaz HF, Hernández-Aguilar M, López-Hernández GR, Zamudio GS, José MV. The Spike Protein of SARS-CoV-2 Is Adapting Because of Selective Pressures. Vaccines. 2022; 10(6):864. https://doi.org/10.3390/vaccines10060864
Chicago/Turabian StyleLópez-Cortés, Georgina I., Miryam Palacios-Pérez, Hannya F. Veledíaz, Margarita Hernández-Aguilar, Gerardo R. López-Hernández, Gabriel S. Zamudio, and Marco V. José. 2022. "The Spike Protein of SARS-CoV-2 Is Adapting Because of Selective Pressures" Vaccines 10, no. 6: 864. https://doi.org/10.3390/vaccines10060864