
Antiviral Efficacy of Selected Natural Phytochemicals against SARS-CoV-2 Spike Glycoprotein Using Structure-Based Drug Designing



Abstract
:1. Introduction
2. Results
2.1. Structure Retrieval of Spike Protein
2.2. Database Screening and Docking Study
2.3. Drug-Likeness/ADMET Profiling
2.4. Biochemical Classification of Idetified Compounds
2.5. Energy Calculations
2.6. MD Simulation
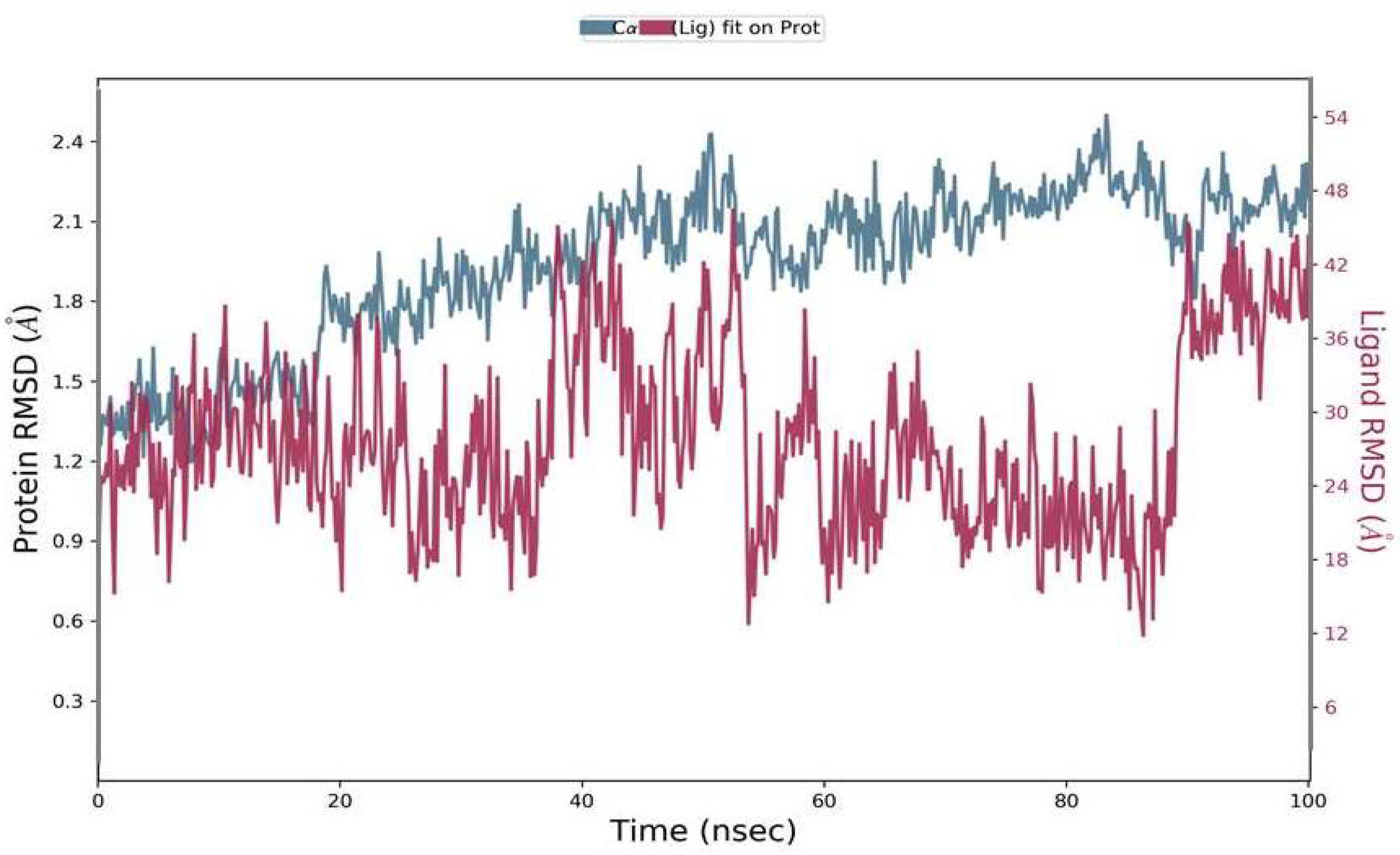
2.6.1. Root Mean Square Deviation (RMSD)
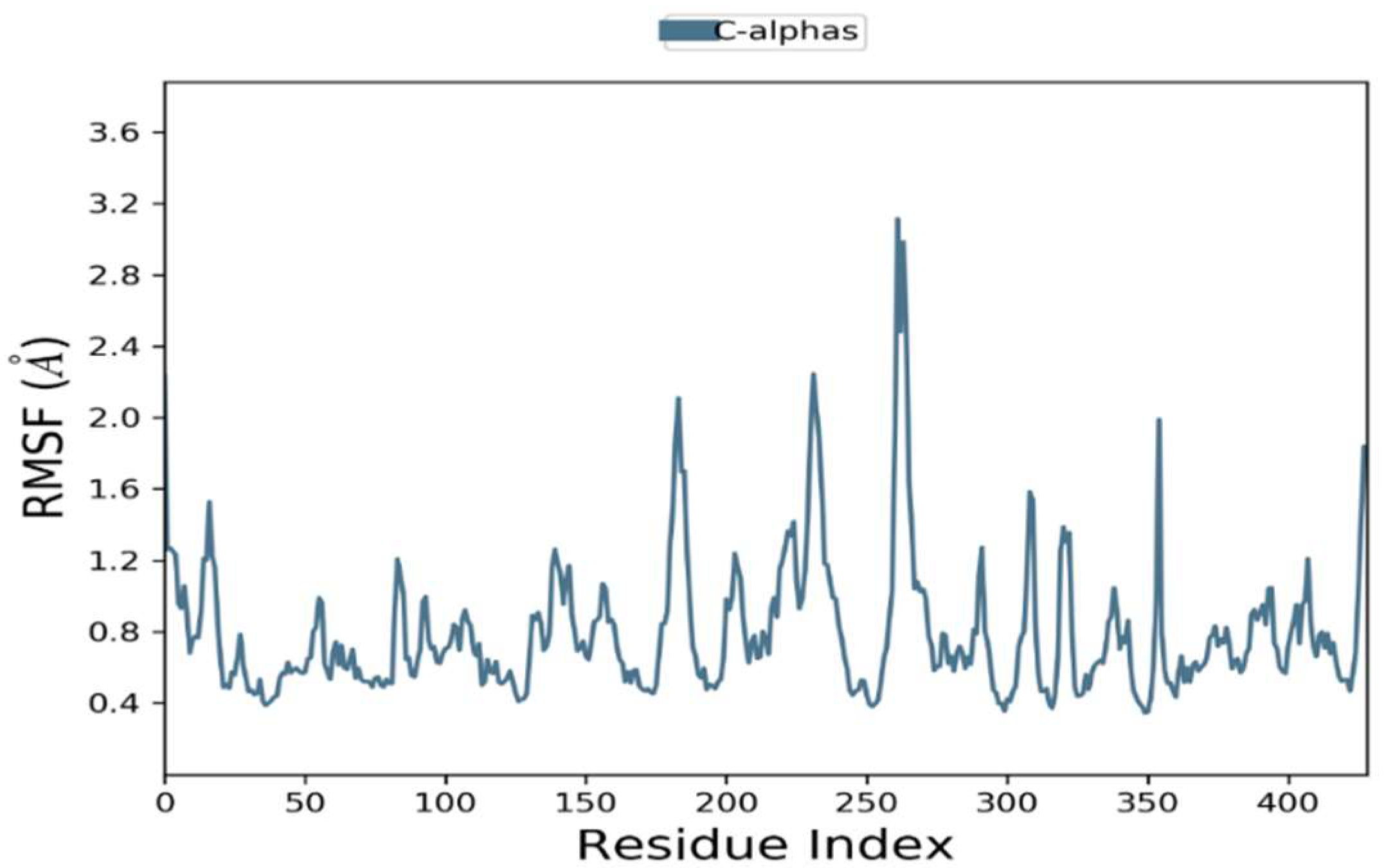
2.6.2. Root Mean Square Fluctuations
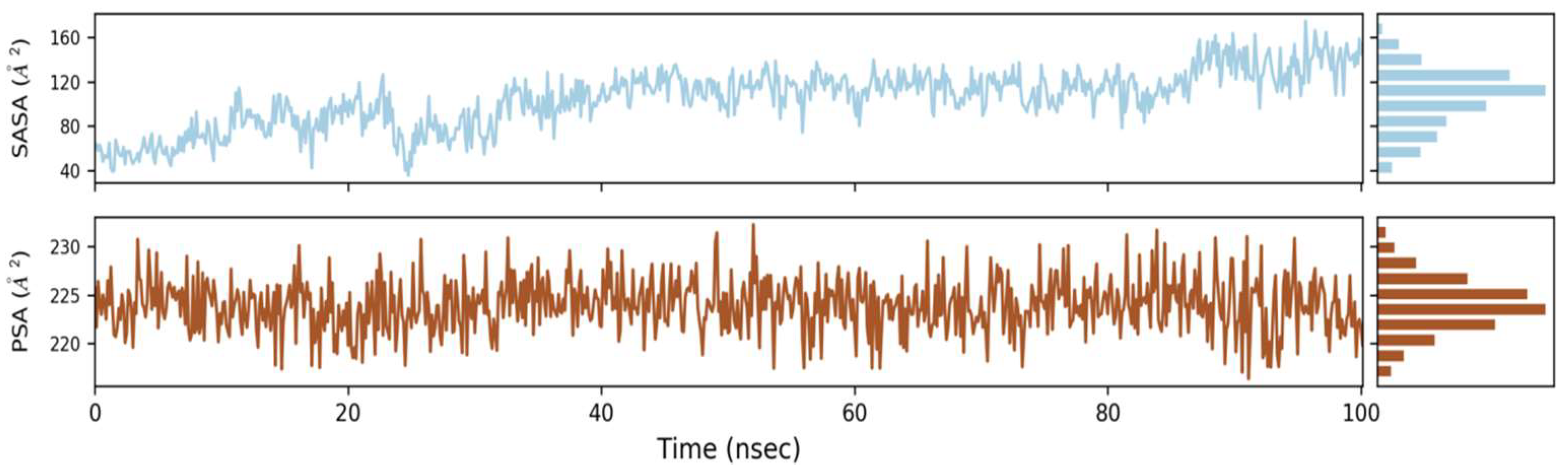
2.6.3. Solvent Accessible Surface Area (SASA)
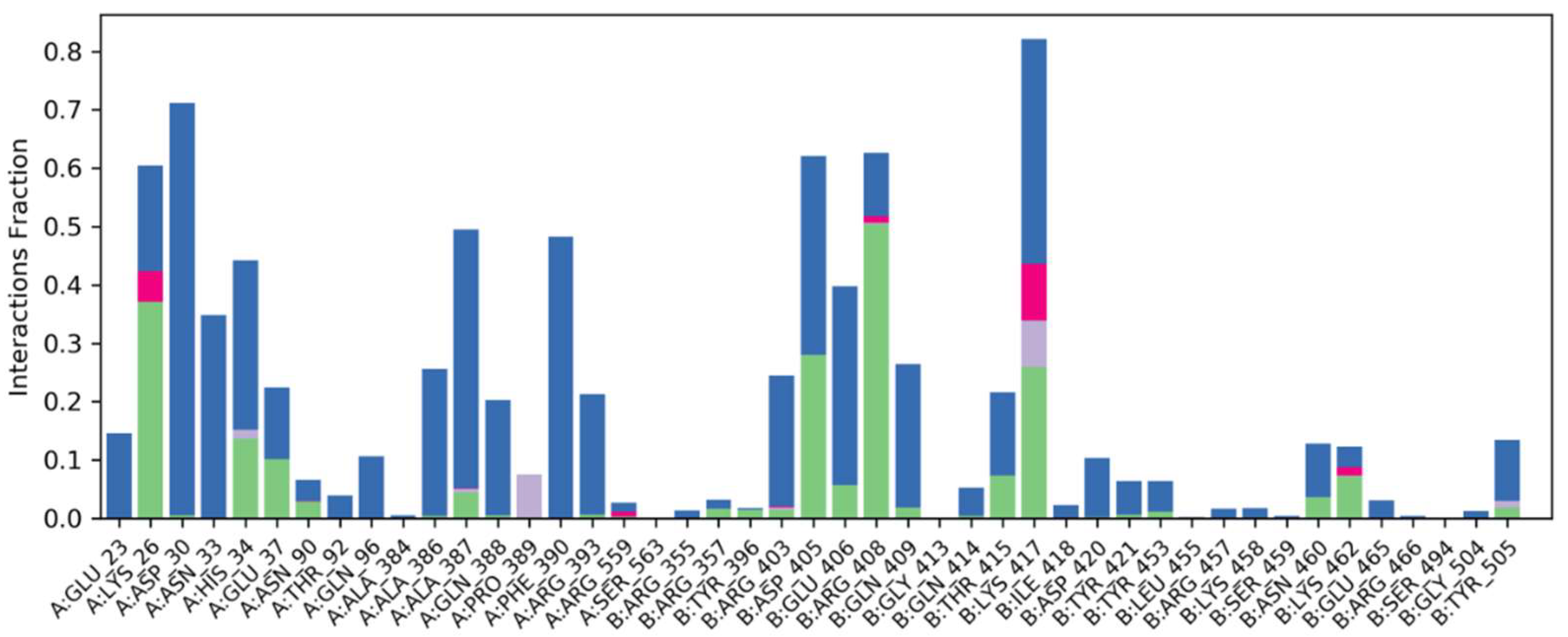
2.6.4. Water Bridges, Ionic Interactions and Hydrogen Bonding Graphs
3. Discussion
4. Materials and Methods
4.1. Data Collection and Ligand Database
4.2. Receptor Preparation and Analysis of Target Active Binding Sites
4.3. Molecular Docking
4.4. Analysis of Ligand Receptor Interaction
4.5. Physiochemical Property Profile and Toxicity Prediction
4.6. MM-GBSA Binding Free Energy Calculations
4.7. Molecular Dynamic Simulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosten, C.; Gunther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.S.; Zheng, B.J.; Li, Y.M.; Poon, L.L.M.; Xie, Z.H.; Chan, K.H.; Li, P.H.; Tan, S.Y.; Chang, Q.; Xie, J.P.; et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef] [Green Version]
- Nosratabadi, R.; Alavian, S.M.; Zare-Bidaki, M.; Shahrokhi, V.M.; Arababadi, M.K. Innate immunity related pathogen recognition receptors and chronic hepatitis B infection. Mol. Immunol. 2017, 90, 64–73. [Google Scholar] [CrossRef]
- Quesada, J.A.; Lopez-Pineda, A.; Gil-Guillen, V.F.; Arriero-Marin, J.M.; Gutierrez, F.; Carratala-Munuera, C. Incubation period of COVID-19: A systematic review and meta-analysis. Periodo de incubacion de la COVID-19: Revision sistematica y metaanalisis. Rev. Clin. Esp. 2021, 221, 109–117. [Google Scholar] [CrossRef]
- Ruiz-Frutos, C.; Ortega-Moreno, M.; Dias, A.; Bernardes, J.M.; Garcia-Iglesias, J.J.; Gomez-Salgado, J. Information on COVID-19 and Psychological Distress in a Sample of Non-Health Workers during the Pandemic Period. Int. J. Environ. Res. Public Health 2020, 17, 6982. [Google Scholar] [CrossRef]
- Brito, C.A.A.; Brito, M.C.M.; Martins, T.H.F.; Brito, C.C.M.; Albuquerque, M.F.M.; Brito, R. Clinical laboratory and dispersion pattern of COVID-19 in a family cluster in the social-distancing period. J. Infect. Dev. Ctries 2020, 14, 987–993. [Google Scholar] [CrossRef]
- Sytar, O.; Brestic, M.; Hajihashemi, S.; Skalicky, M.; Jan, K.; Lamilla-Tamayo, L.; Ibrahimova, U.; Ibadullayeva, S.; Landi, M. COVID-19 prophylaxis efforts based on natural antiviral plant extracts and their compounds. Molecules 2021, 26, 727. [Google Scholar] [CrossRef]
- Kossel, A. Ueber die chemische Zusammensetzung der Zelle. Du Bois-Reymond’s Arch./Arch. Anat. Physiol. Physiol. Abt. 1891, 278, 181–186. [Google Scholar]
- Abiri, R.; Abdul-Hamid, H.; Sytar, O.; Abiri, R.; Bezerra de Almeida, E.; Sharma, S.K.; Bulgakov, V.P.; Arroo, R.R.J.; Malik, S. A Brief Overview of Potential Treatments for Viral Diseases Using Natural Plant Compounds: The Case of SARS-Cov. Molecules 2021, 26, 3868. [Google Scholar] [CrossRef]
- Moline, J.; Bukharovich, I.; Wolff, M.; Phillips, R. Dietary flavonoids and hypertension: Is there a link? Med. Hypotheses 2000, 55, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Soleymani, S.; Naghizadeh, A.; Karimi, M.; Zarei, A.; Mardi, R.; Kordafshari, G.; Esmaealzadeh, N.; Zargaran, A. COVID-19: General Strategies for Herbal Therapies. J. Evid.-Based Integr. Med. 2022, 27, 2515690X211053641. [Google Scholar] [CrossRef] [PubMed]
- Nyamai, D.W.; Arika, W.; Ogola, P.; Njagi, E.; Ngugi, M. Medicinally important phytochemicals: An untapped research avenue. J. Pharmacogn. Phytochem. 2016, 4, 2321–6182. [Google Scholar]
- John, B.; Sulaiman, C.; George, S.; Reddy, V. Total phenolics and flavonoids in selected medicinal plants from Kerala. Int. J. Pharm. Pharm. Sci. 2014, 6, 406–408. [Google Scholar]
- Chiavaroli, V.; Giannini, C.; De Marco, S.; Chiarelli, F.; Mohn, A. Unbalanced oxidant–antioxidant status and its effects in pediatric diseases. Redox Rep. 2011, 16, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Ji, M.; Sikandar, A.; Iram, A.; Qin, P.; Zhu, H.; Javeed, A.; Shafi, J.; Iqbal, Z.; Iqbal, M.F.; et al. Phytochemical Analysis, Biochemical and Mineral Composition and GC-MS Profiling of Methanolic Extract of Chinese Arrowhead Sagittaria trifolia L. from Northeast China. Molecules 2019, 24, 3025. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.-I.; Matsui, T. Nitric oxide, a janus-faced therapeutic target for diabetic microangiopathy—Friend or foe? Pharmacol. Res. 2011, 64, 187–194. [Google Scholar] [CrossRef]
- Carfì, A.; Bernabei, R.; Landi, F. Persistent symptoms in patients after acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Li, X.-L.; Yan, Z.-R.; Sun, X.-P.; Han, J.; Zhang, B.-W. Potential neurological symptoms of COVID-19. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420917830. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Han, B.; Wang, J. COVID-19: Gastrointestinal manifestations and potential fecal–oral transmission. Gastroenterology 2020, 158, 1518–1519. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Lian, J.S.; Hu, J.H.; Gao, J.; Zheng, L.; Zhang, Y.M.; Hao, S.R.; Jia, H.Y.; Cai, H.; Zhang, X.L.; et al. Epidemiological, clinical and virological characteristics of 74 cases of coronavirus-infected disease 2019 (COVID-19) with gastrointestinal symptoms. Gut 2020, 69, 1002–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struyf, T.; Deeks, J.J.; Dinnes, J.; Takwoingi, Y.; Davenport, C.; Leeflang, M.M.; Spijker, R.; Hooft, L.; Emperador, D.; Dittrich, S.; et al. Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19 disease. Cochrane Database Syst. Rev. 2020, 7, CD013665. [Google Scholar] [PubMed]
- Kolifarhood, G.; Aghaali, M.; Saadati, H.M.; Taherpour, N.; Rahimi, S.; Izadi, N.; Nazari, S.S.H. Epidemiological and clinical aspects of COVID-19; a narrative review. Arch. Acad. Emerg. Med. 2020, 8, e41. [Google Scholar] [PubMed]
- Saad, M.; Omrani, A.; Baig, K.; Bahloul, A.; Elzein, F.; Matin, M.A.; Selim, M.A.; Al Mutairi, M.; Al Nakhli, D.; Al Aidaroos, A.Y.; et al. Clinical aspects and outcomes of 70 patients with Middle East respiratory syndrome coronavirus infection: A single-center experience in Saudi Arabia. Int. J. Infect. Dis. 2014, 29, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Mourier, T.; Shuaib, M.; Hala, S.; Mfarrej, S.; Alofi, F.; Naeem, R.; Alsomali, A.; Jorgensen, D.; Subudhi, A.K.; Rached, F.B.; et al. SARS-CoV-2 genomes from Saudi Arabia implicate nucleocapsid mutations in host response and increased viral load. Nat. Commun. 2022, 13, 601. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): Emergence, history, basic and clinical aspects. Saudi J. Biol. Sci. 2020, 27, 2531–2538. [Google Scholar] [CrossRef]
- Marshall, G.R. Computer-aided drug design. Annu. Rev. Pharmacol. Toxicol. 1987, 27, 193–213. [Google Scholar] [CrossRef]
- Kapetanovic, I. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem.-Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, S.; Malik, Y.S.; Tomar, S. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front. Immunol. 2020, 11, 1664. [Google Scholar] [CrossRef]
- Paredes-Ramos, M.; Conde Piñeiro, E.; Pérez-Sánchez, H.; López-Vilariño, J.M. Evaluation of Natural Peptides to Prevent and Reduce the Novel SARS-CoV-2 Infection. J. Food Qual. 2022, 2022, 2102937. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Pei, S.; Chen, B.; Song, Y.; Zhang, T.; Yang, W.; Shaman, J. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science 2020, 368, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Hanai, T. Quantitative in silico analysis of SARS-CoV-2 S-RBD omicron mutant transmissibility. Talanta 2022, 240, 123206. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Mudgal, R.; Nehul, S.; Tomar, S. Prospects for mucosal vaccine: Shutting the door on SARS-CoV-2. Hum. Vaccines Immunother. 2020, 16, 2921–2931. [Google Scholar] [CrossRef]
- Adedeji, A.O.; Severson, W.; Jonsson, C.; Singh, K.; Weiss, S.R.; Sarafianos, S.G. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J. Virol. 2013, 87, 8017–8028. [Google Scholar] [CrossRef] [Green Version]
- Ashfaq, U.A.; Mumtaz, A.; ul Qamar, T.; Fatima, T. MAPS Database: Medicinal plant activities, phytochemical and structural database. Bioinformation 2013, 9, 993. [Google Scholar] [CrossRef]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated platform of small molecules and biological activities. Annu. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Modeling 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumtaz, A.; Ashfaq, U.A.; Qamar, M.T.U.; Anwar, F.; Gulzar, F.; Ali, M.A.; Saari, N.; Pervez, M.T. MPD3: A useful medicinal plants database for drug designing. Nat. Prod. Res. 2017, 31, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Mangal, M.; Sagar, P.; Singh, H.; Raghava, G.P.; Agarwal, S.M. NPACT: Naturally occurring plant-based anti-cancer compound-activity-target database. Nucleic Acids Res. 2013, 41, D1124–D1129. [Google Scholar] [CrossRef] [Green Version]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Yuriev, E.; Agostino, M.; Ramsland, P.A. Challenges and advances in computational docking: 2009 in review. J. Mol. Recognit. 2011, 24, 149–164. [Google Scholar] [CrossRef]
- Moitessier, N.; Englebienne, P.; Lee, D.; Lawandi, J.; Corbeil, C.R. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharmacol. 2008, 153, S7–S26. [Google Scholar] [CrossRef] [Green Version]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. eHiTS: A new fast, exhaustive flexible ligand docking system. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein–ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef] [PubMed]
- Inc. CCG. Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2016. [Google Scholar]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Al-Khafaji, K.; Al-Duhaidahawi, D.; Tok, T.T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory. Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IDs (PubChem) | Phytochemicals | Binding Affinity (kcal/mol) | RMSD Value | Hydrogen Bonds and Other Interacting Residues |
|---|---|---|---|---|
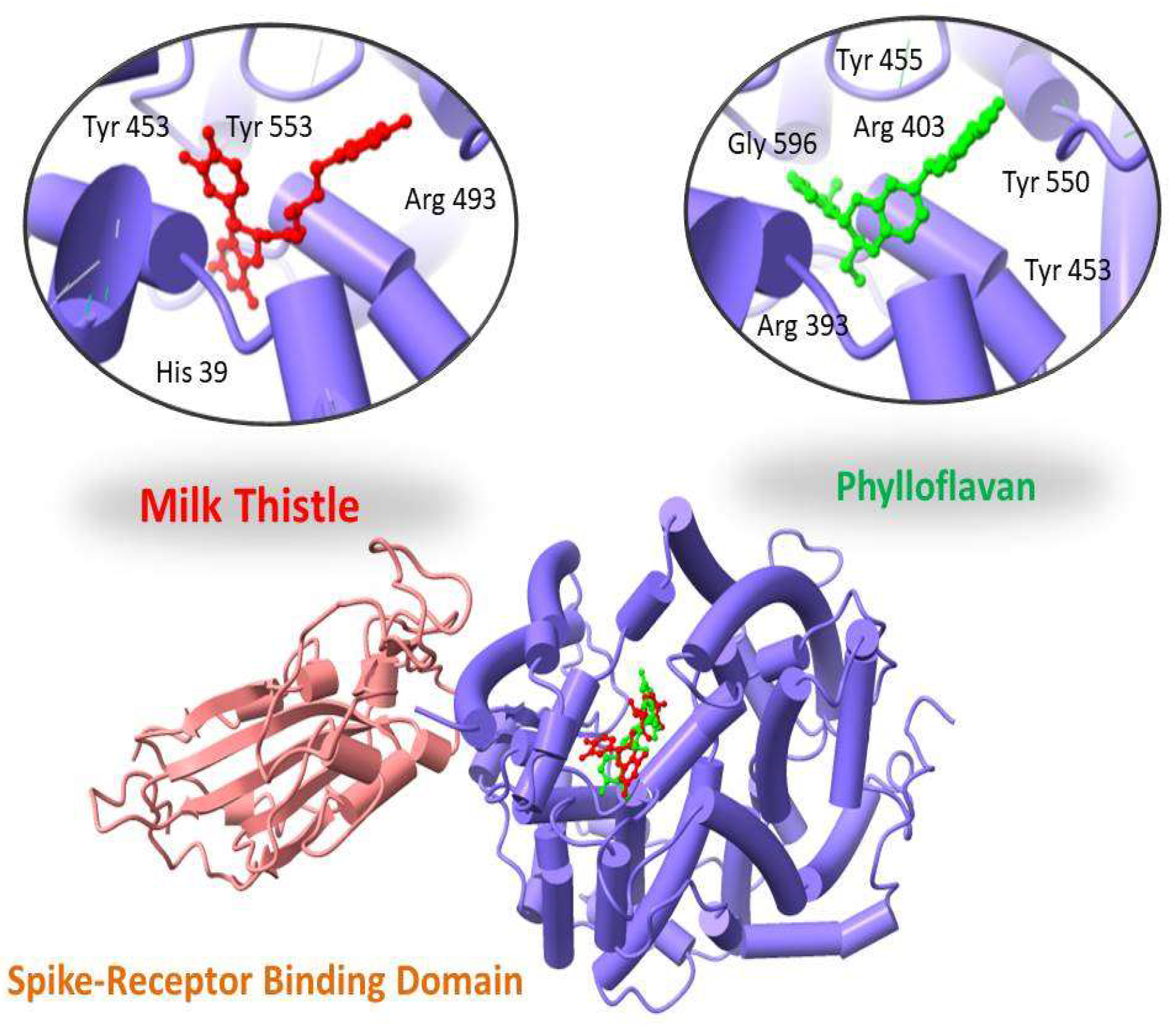
| 457885 | Phylloflavan | −14.09 | 0.59 | Tyr 455; Tyr 550; Arg 393; Gly 596; Tyr 453; Arg 403 |
| 1548994 | Milk thistle | −13.10 | 1.01 | Tyr 453; Tyr 553; Arg 493; His 39 |
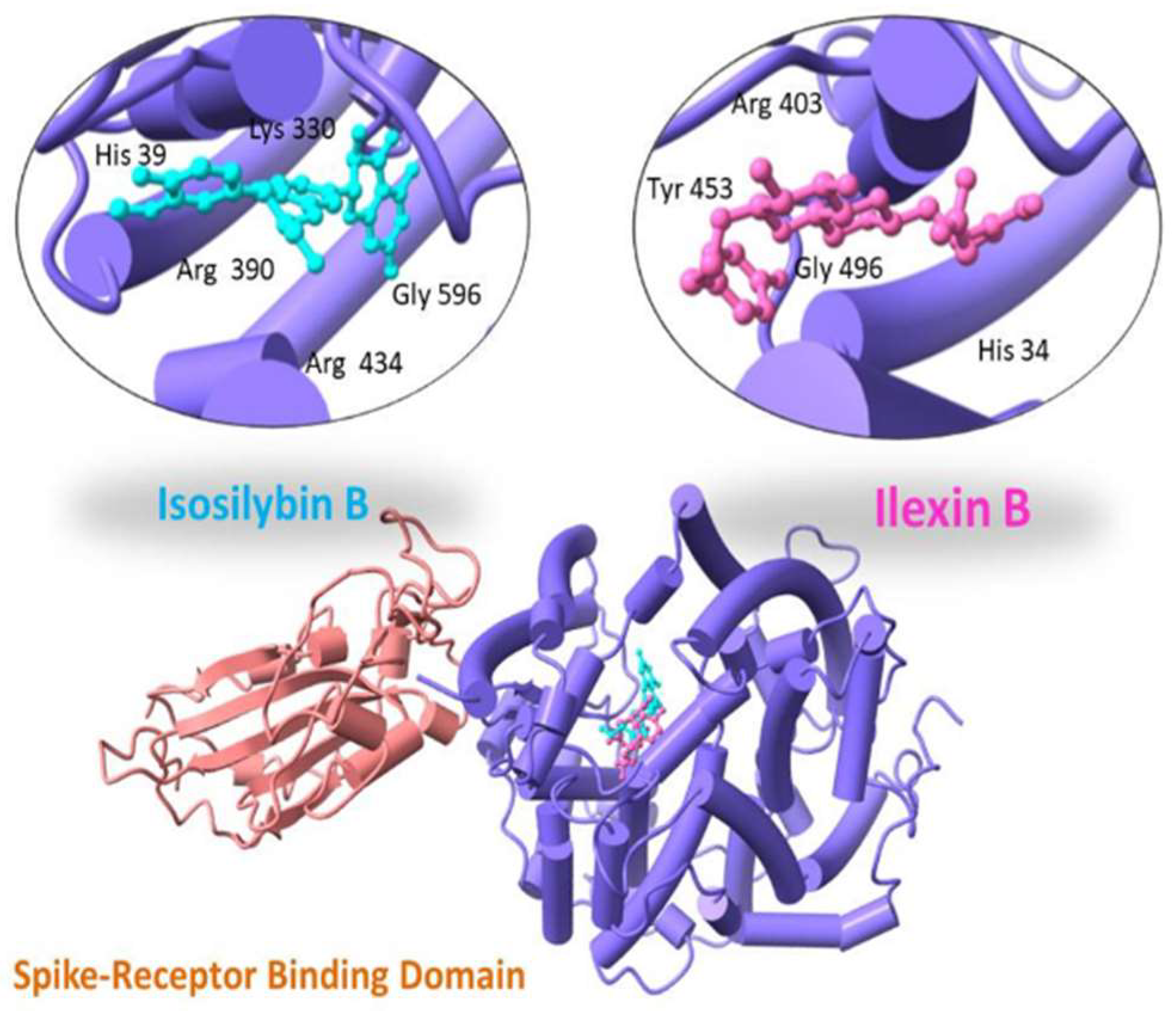
| 102394711 | Ilexin B | −13.04 | 1.53 | Tyr 453; Gly 496; His 34; Arg 403 |
| 10885340 | Isosilybin B | −12.19 | 0.92 | Arg 390; Arg 434; Lys 330; His 39 Gly 596 |
| Sr No | Compounds Name | Log P | M Weight | HBD | HBA |
|---|---|---|---|---|---|
| 1 | Phylloflavan | 2.53 | 498.48 | 10 | 4 |
| 2 | Milk thistle | −1.25 | 481.43 | 10 | 4 |
| 3 | Ilexin B | −1.82 | 464 | 10 | 2 |
| 4 | Isosilybin B | −1.25 | 481.43 | 10 | 4 |
| Compounds | Phylloflavan | Milk Thistle | Ilexin B | Isosilybin B |
|---|---|---|---|---|
| Absorption | ||||
| Blood-Brain Barrier | No | No | No | No |
| Distribution | ||||
| Gastro-Intestinal-Absorption | Low | Low | Low | Low |
| P-glycoprotein-substrate | No | No | Yes | No |
| CYP450-1A2-Inhibitor | No | No | No | No |
| Metabolisum | ||||
| CYP450-2C9-Inhibitor | No | No | No | No |
| CYP450-2D6-Inhibitor | No | No | No | No |
| CYP450-2C19-Inhibitor | No | No | Yes | No |
| CYP450-3A4 Inhibitor | No | No | No | No |
| Toxicity | ||||
| Cytotoxicity | Non-toxic | Non-toxic | Non-toxic | Non-toxic |
| Immunogenicity | Non-toxic | Non-toxic | Non-toxic | Non-toxic |
| Mutagenicity | Non-toxic | Non-toxic | Non-toxic | Non-toxic |
| Compounds | Taxonomy | Classification | Diseases |
|---|---|---|---|
| Phylloflavan | Phyllocladus trichomanoides Phyllocladus alpinus | Polyketides Flavonoids Flavans, Flavanols and Leucoanthocyanidins | Antileishmanial activity and modulatory effects on nitric oxide and tumor necrosis human immunodeficiency virus type 1 integrase |
| Milk thistle | Anastatica hierochuntica Silybum marianum Aspergillus iizukae | Flavonoids silibinin dehydrosilibinin silychristin silydianin | Liver disorders and gallbladder problems.hepatitis, cirrhosis, jaundice, diabetes, indigestion |
| Ilexin B | Panax notoginseng | Glucosides Carbohydrates | Inflammatory bowel disease, arthritis, ischemia, atherosclerosis, Alzheimer disease and trauma, as well as hyperlipidemia, diabetes |
| Isosilybin B | Anastatica hierochuntica Silybum marianum | Hydrocarbons, Aromatic Hydrocarbons, Cyclic Benzene Derivatives Flavonolignans | Antiprostate cancer activity via inhibiting proliferation and inducing G1 phase arrestand apoptosia. |
| Energy Parameters | VDWAALS (kcal mol−1) | Delta G Gas (kcal mol−1) | Delta g Solv (kcal mol−1) | Delta Total (kcal mol−1) |
|---|---|---|---|---|
| Phylloflavan/S-RBD | −29.50 | −34.56 | 8.21 | −30.35 |
| Milk thistle/S-RBD | −30.61 | −31.87 | 9.86 | −28.90 |
| Ilexin B/S-RBD | −31.70 | −29.54 | 10.23 | −31.83 |
| Isosilybin B/S-RBD | −28.74 | −32.33 | 11.23 | −34.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aloufi, B.H.; Snoussi, M.; Sulieman, A.M.E. Antiviral Efficacy of Selected Natural Phytochemicals against SARS-CoV-2 Spike Glycoprotein Using Structure-Based Drug Designing. Molecules 2022, 27, 2401. https://doi.org/10.3390/molecules27082401
Aloufi BH, Snoussi M, Sulieman AME. Antiviral Efficacy of Selected Natural Phytochemicals against SARS-CoV-2 Spike Glycoprotein Using Structure-Based Drug Designing. Molecules. 2022; 27(8):2401. https://doi.org/10.3390/molecules27082401
Chicago/Turabian StyleAloufi, Bandar Hamad, Mejdi Snoussi, and Abdel Moneim E. Sulieman. 2022. "Antiviral Efficacy of Selected Natural Phytochemicals against SARS-CoV-2 Spike Glycoprotein Using Structure-Based Drug Designing" Molecules 27, no. 8: 2401. https://doi.org/10.3390/molecules27082401