
Electrostatic Map of the SARS-CoV-2 Virion Specifies Binding Sites of the Antiviral Cationic Photosensitizer

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
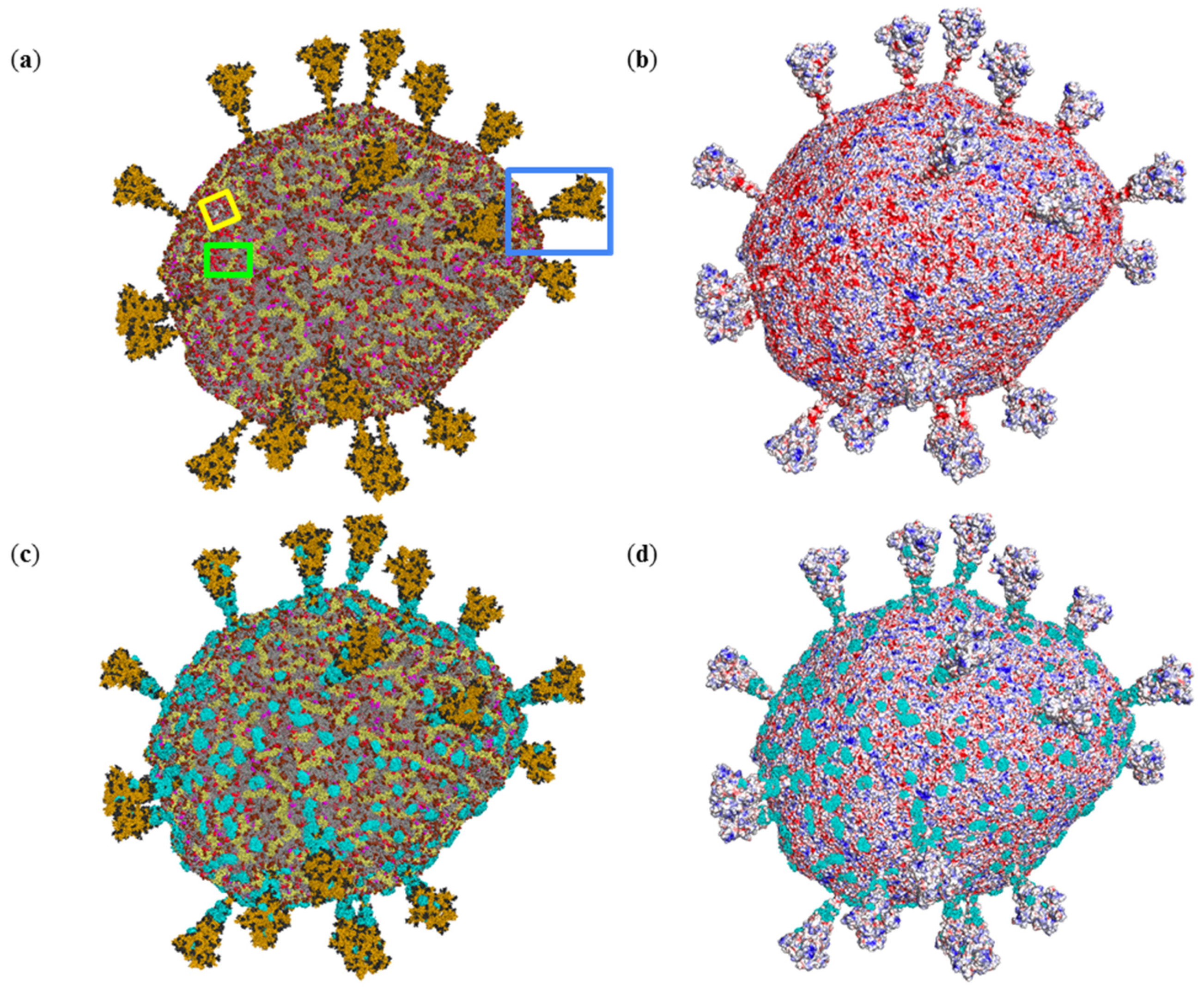
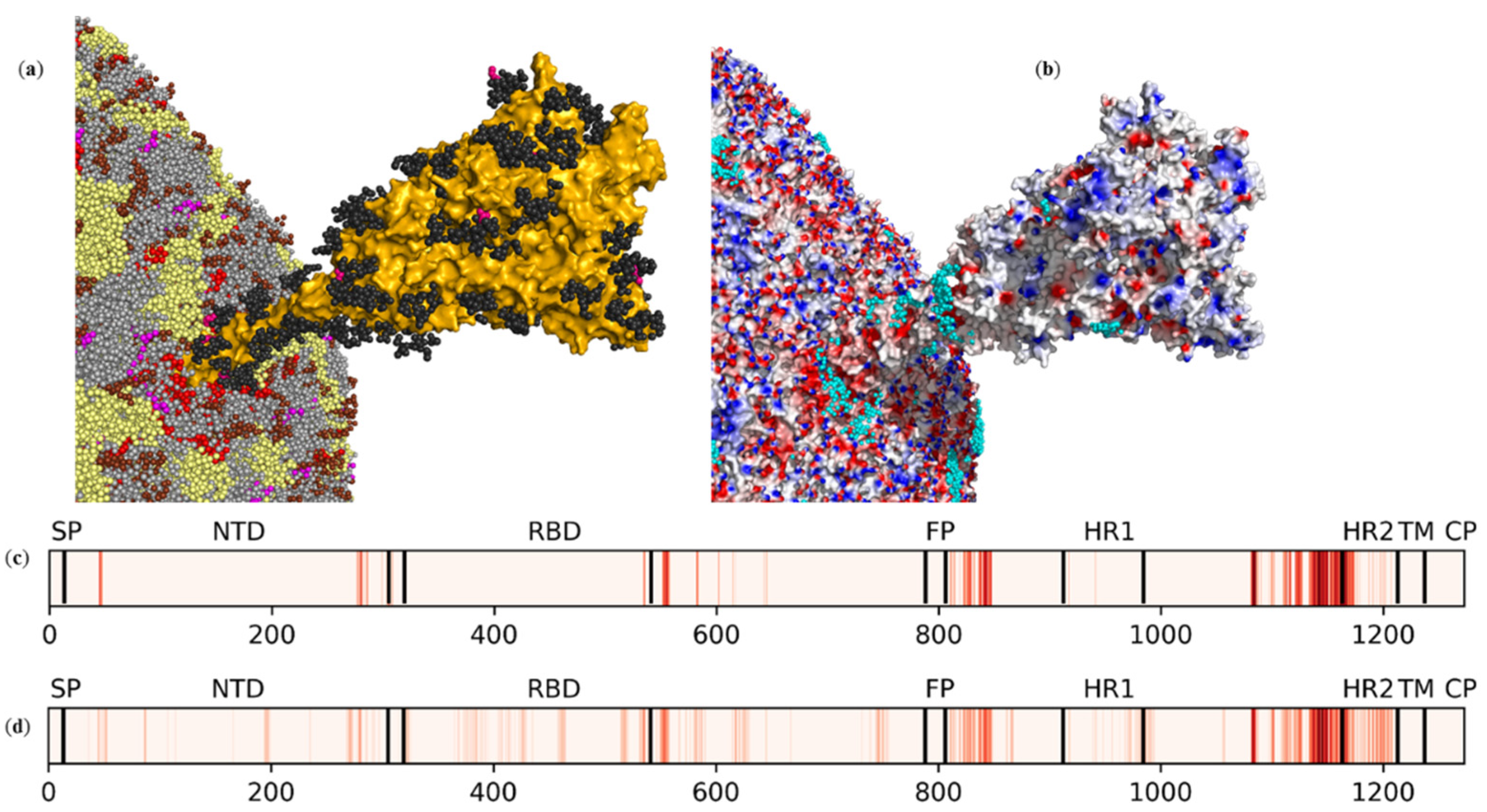
2.1. Electrostatic Potential Distribution of the SARS-CoV-2 Envelope
2.2. Interactions of Photosensitizer with the SARS-CoV-2 Envelope Revealed by Brownian Dynamics
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zmasek, C.M.; Lefkowitz, E.J.; Niewiadomska, A.; Scheuermann, R.H. Genomic evolution of the Coronaviridae family. Virology 2022, 570, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2021, 54, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R.R.; Carpenter, A.; Liew, A.Y.; Martin, K.B.; Herring, M.K.; Gerber, S.I.; Hall, A.J.; Sleeman, J.M.; VonDobschuetz, S.; Behravesh, C.B. Animal reservoirs and hosts for emerging alphacoronaviruses and betacoronaviruses. Emerg. Infect. Dis. 2021, 27, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, F.K. The proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T. Viewing SARS-CoV-2 nucleocapsid protein in terms of molecular flexibility. Biology 2021, 10, 454. [Google Scholar] [CrossRef]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738.e13. [Google Scholar] [CrossRef]
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.M.; et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef]
- Van Meer, G.; de Kroon, A.I. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124 Pt 1, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef]
- Barrantes, F.J. Structural biology of coronavirus ion channels. Acta Crystallogr. D Struct. Biol. 2021, 77 Pt 4, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Ruch, T.R.; Machamer, C.E. The coronavirus E protein: Assembly and beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef] [Green Version]
- Verdiá-Báguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; DeDiego, M.L.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology 2012, 432, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, R.; Wang, W.; Jiang, S.; Yang, C.; Liu, N.; Dai, H.; Lee, I.; Meng, X.; Yuan, Z. Probing the formation, structure and free energy relationships of M protein dimers of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 573–582. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Barrantes, F.J. The contribution of biophysics and structural biology to current advances in COVID-19. Annu. Rev. Biophys. 2021, 50, 493–523. [Google Scholar] [CrossRef]
- Bárcena, M.; Barnes, C.O.; Beck, M.; Bjorkman, P.J.; Canard, B.; Gao, G.F.; Gao, Y.; Hilgenfeld, R.; Hummer, G.; Patwardhan, A.; et al. Structural biology in the fight against COVID-19. Nat. Struct. Mol. Biol. 2021, 28, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Li, S. Cryo-electron tomography of enveloped viruses. Trends Biochem. Sci. 2022, 47, 173–186. [Google Scholar] [CrossRef]
- Ingólfsson, H.I.; Arnarez, C.; Periole, X.; Marrink, S.J. Computational ‘microscopy’ of cellular membranes. J. Cell Sci. 2016, 129, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Arkhipov, A.; Freddolino, P.L.; Schulten, K. Stability and dynamics of virus capsids described by coarse-grained modeling. Structure 2006, 14, 1767–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Jung, J.; Kobayashi, C.; Torre, D.U.; Takada, S.; Sugita, Y. Implementation of residue-level coarse-grained models in GENESIS for large-scale molecular dynamics simulations. PLoS Comput. Biol. 2022, 18, e1009578. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.; Shorthouse, D.; Parton, D.L.; Jefferys, E.; Fowler, P.W.; Chavent, M.; Baaden, M.; Sansom, M.S.P. Nothing to sneeze at: A dynamic and integrative computational model of an influenza A virion. Structure 2015, 23, 584–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, T.; Sansom, M.S. The role of the membrane in the structure and biophysical robustness of the dengue virion envelope. Structure 2016, 24, 375–382. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhong, C.; Tieleman, D.P. Supramolecular organization of SARS-CoV and SARS-CoV-2 virions revealed by coarse-grained models of intact virus envelopes. J. Chem. Inf. Model. 2022, 62, 176–186. [Google Scholar] [CrossRef]
- Yu, A.; Pak, A.J.; He, P.; Monje-Galvan, V.; Casalino, L.; Gaieb, Z.; Dommer, A.C.; Amaro, R.E.; Voth, G.A. A multiscale coarse-grained model of the SARS-CoV-2 virion. Biophys. J. 2021, 120, 1097–1104. [Google Scholar] [CrossRef]
- Pezeshkian, W.; Grünewald, F.; Narykov, O.; Lu, S.; Wassenaar, T.A.; Marrink, S.J.; Korkin, D. Molecular architecture of SARS-CoV-2 envelope by integrative modeling. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhukhovitsky, V.; Shevlyagina, N.; Zubasheva, M.; Russu, L.; Gushchin, V.; Meerovich, G.; Strakhovskaya, M. Infectivity and morphology of bovine coronavirus inactivated in vitro by cationic photosensitizers. Viruses 2022, 14, 1053. [Google Scholar] [CrossRef] [PubMed]
- Korneev, D.; Kurskaya, O.; Sharshov, K.; Eastwood, J.; Strakhovskaya, M. Ultrastructural aspects of photodynamic inactivation of highly pathogenic avian H5N8 influenza virus. Viruses 2019, 11, 955. [Google Scholar] [CrossRef] [Green Version]
- Sharshov, K.; Solomatina, M.; Kurskaya, O.; Kovalenko, I.; Kholina, E.; Fedorov, V.; Meerovich, G.; Rubin, A.; Strakhovskaya, M. The photosensitizer octakis(cholinyl)zinc phthalocyanine with ability to bind to a model spike protein leads to a loss of SARS-CoV-2 infectivity in vitro when exposed to far-red LED. Viruses 2021, 13, 643. [Google Scholar] [CrossRef]
- Fedorov, V.; Kholina, E.; Khruschev, S.; Kovalenko, I.; Rubin, A.; Strakhovskaya, M. What binds cationic photosensitizers better: Brownian dynamics reveals key interaction sites on spike proteins of SARS-CoV, MERS-CoV, and SARS-CoV-2. Viruses 2021, 13, 1615. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Du, D.; Karki, C.B.; Guo, W.; Lopez-Hernandez, A.E.; Sun, S.; Juarez, B.Y.; Li, H.; Wang, J.; Li, L. Revealing the mechanism of SARS-CoV-2 spike protein binding with ACE2. Comput. Sci. Eng. 2020, 22, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, S.; Ciccozzi, M.; Zella, D.; Bianchi, M.; Benedetti, F.; Benvenuto, D.; Broccolo, F.; Cauda, R.; Caruso, A.; Angeletti, S.; et al. SARS-CoV-2 B.1.617 Indian variants: Are electrostatic potential changes responsible for a higher transmission rate? J. Med. Virol. 2021, 93, 6551–6556. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, S.; Ciccozzi, M.; Bianchi, M.; Benvenuto, D.; Cauda, R.; Cassone, A. The value of electrostatic potentials of the spike receptor binding and N-terminal domains in addressing transmissibility and infectivity of SARS-CoV-2 variants of concern. J. Infect. 2022, 84, e62–e63. [Google Scholar] [CrossRef] [PubMed]
- Heffron, J.; Mayer, B.K. Virus isoelectric point estimation: Theories and methods. Appl. Environ. Microbiol. 2021, 87, e02319-20. [Google Scholar] [CrossRef]
- Michen, B.; Graule, T. Isoelectric points of viruses. J. Appl. Microbiol. 2010, 109, 388–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Areo, O.; Joshi, P.U.; Obrenovich, M.; Tayahi, M.; Heldt, C.L. Single-particle characterization of SARS-CoV-2 isoelectric point and comparison to variants of interest. Microorganisms 2021, 9, 1606. [Google Scholar] [CrossRef]
- Scheller, C.; Krebs, F.; Minkner, R.; Astner, I.; Gil-Moles, M.; Wätzig, H. Physicochemical properties of SARS-CoV-2 for drug targeting, virus inactivation and attenuation, vaccine formulation and quality control. Electrophoresis 2020, 41, 1137–1151. [Google Scholar] [CrossRef]
- Ghatak, S.; Khona, D.K.; Sen, A.; Huang, K.; Jagdale, G.; Singh, K.; Gopalakrishnan, V.; Cornetta, K.G.; Roy, S.; Khanna, S.; et al. Electroceutical fabric lowers zeta potential and eradicates coronavirus infectivity upon contact. Sci. Rep. 2021, 11, 21723. [Google Scholar] [CrossRef]
- Marrink, S.J.; Tieleman, D.P. Perspective on the Martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, P.S.; Kholina, E.G.; Bozdaganyan, M.E.; Nesterenko, A.M.; Kovalenko, I.B.; Strakhovskaya, M.G. Molecular mechanism of uptake of cationic photoantimicrobial phthalocyanine across bacterial membranes revealed by molecular dynamics simulations. J. Phys. Chem. B 2018, 122, 3711–3722. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, I.B.; Khrushchev, S.S.; Fedorov, V.A.; Riznichenko, G.Y.; Rubin, A.B. The role of electrostatic interactions in the process of diffusional encounter and docking of electron transport proteins. Dokl. Biochem. Biophys. 2016, 468, 183–186. [Google Scholar] [CrossRef]
- Fedorov, V.A.; Kovalenko, I.B.; Khruschev, S.S.; Ustinin, D.M.; Antal, T.K.; Riznichenko, G.Y.; Rubin, A.B. Comparative analysis of plastocyanin- cytochrome f complex formation in higher plants, green algae and cyanobacteria. Physiol. Plant. 2019, 166, 320–335. [Google Scholar] [CrossRef] [PubMed]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Dotson, D.L.; Domanski, J.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; Benthall, S., Rostrup, S., Eds.; SciPy: Austin, TX, USA, 2016; pp. 98–105. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L.L.C.; DeLano, W. The PyMOL Molecular Graphics System, Version 2.5. Available online: https://pymol.org/ (accessed on 3 June 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components of the Viral Envelope | Fraction, % | ||
|---|---|---|---|
| proteins | S | 33.9 | |
| M | 8.4 | ||
| E | 0.6 | ||
| lipids | negatively charged | POPI | 43.9 |
| CDL2 | 40.6 | ||
| POPS | 11.5 | ||
| uncharged | POPC | 61.1 | |
| POPE | 39.4 | ||
| CHOL | 0.5 | ||
| Number of Lipid Partners of ZnPcChol8+ | POPI | POPS | CDL2 | Fraction, % |
|---|---|---|---|---|
| 1 | 0 | 0 | 1 | 18 |
| 1 | 0 | 1 | 0 | 3 |
| 1 | 1 | 0 | 0 | 13 |
| 2 | 0 | 1 | 1 | 3 |
| 2 | 0 | 0 | 2 | 7 |
| 2 | 2 | 0 | 0 | 9 |
| 2 | 1 | 1 | 0 | 4 |
| 2 | 1 | 0 | 1 | 19 |
| 3 | 1 | 1 | 1 | 3 |
| 3 | 1 | 0 | 2 | 3 |
| 3 | 2 | 1 | 0 | 2 |
| 3 | 2 | 0 | 1 | 7 |
| 3 | 3 | 0 | 0 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedorov, V.; Kholina, E.; Khruschev, S.; Kovalenko, I.; Rubin, A.; Strakhovskaya, M. Electrostatic Map of the SARS-CoV-2 Virion Specifies Binding Sites of the Antiviral Cationic Photosensitizer. Int. J. Mol. Sci. 2022, 23, 7304. https://doi.org/10.3390/ijms23137304
Fedorov V, Kholina E, Khruschev S, Kovalenko I, Rubin A, Strakhovskaya M. Electrostatic Map of the SARS-CoV-2 Virion Specifies Binding Sites of the Antiviral Cationic Photosensitizer. International Journal of Molecular Sciences. 2022; 23(13):7304. https://doi.org/10.3390/ijms23137304
Chicago/Turabian StyleFedorov, Vladimir, Ekaterina Kholina, Sergei Khruschev, Ilya Kovalenko, Andrew Rubin, and Marina Strakhovskaya. 2022. "Electrostatic Map of the SARS-CoV-2 Virion Specifies Binding Sites of the Antiviral Cationic Photosensitizer" International Journal of Molecular Sciences 23, no. 13: 7304. https://doi.org/10.3390/ijms23137304