Article Text

Statistics from Altmetric.com
Description
A woman in her 40s presented to the eye emergency department with lethargy, headache and blurred vision 2 weeks after receiving her second dose of SARS-CoV-2 mRNA (Comirnaty, Pfizer-BioNTech Manufacturing). Her best-corrected visual acuity (BCVA) was 6/18 and 1/60 in her right and left eyes, respectively. She had no relevant medical history and was a non-smoker.
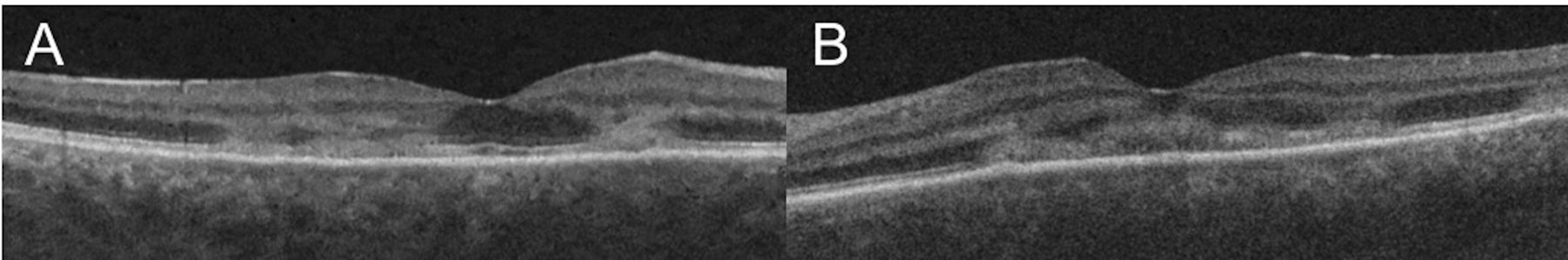
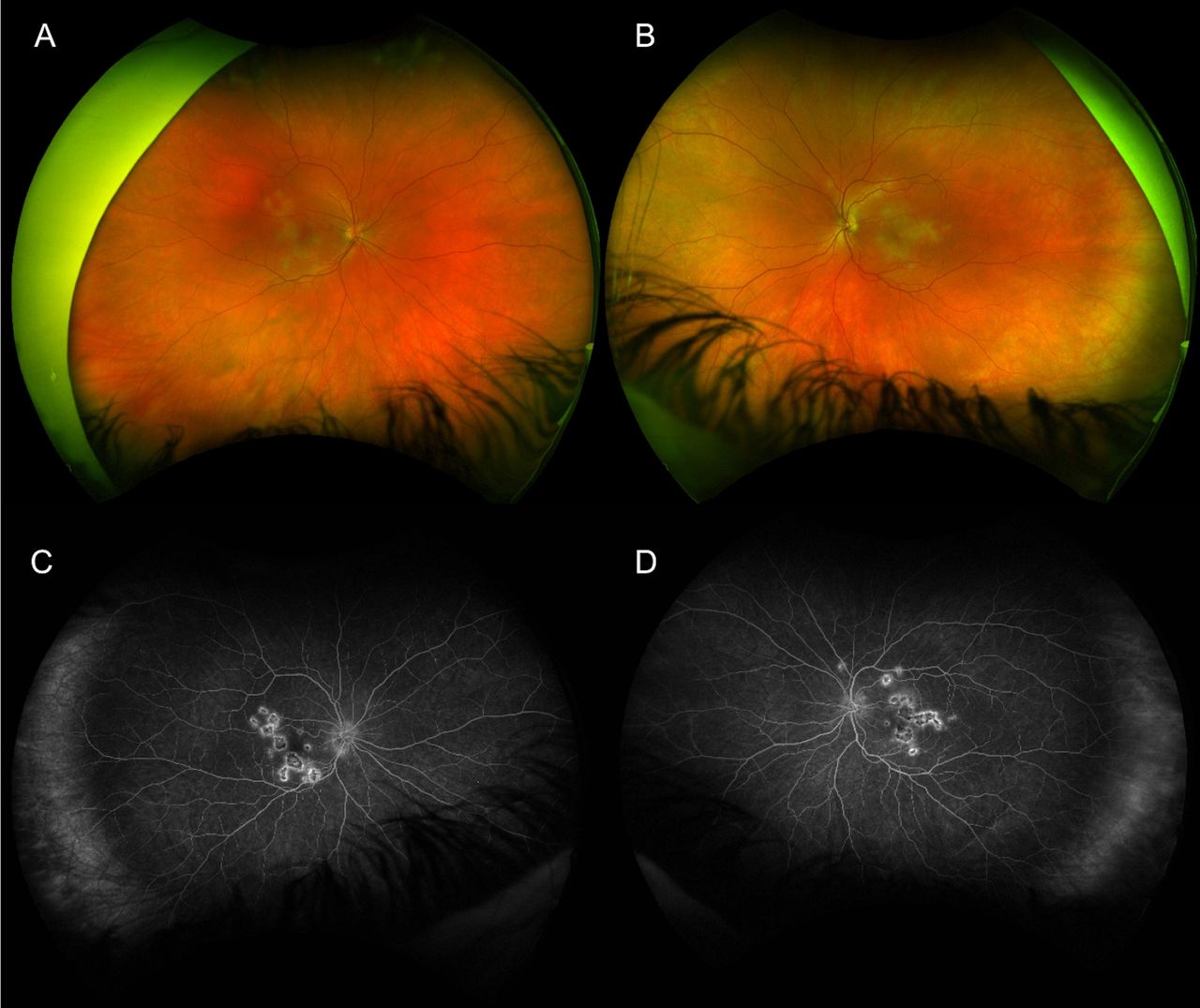
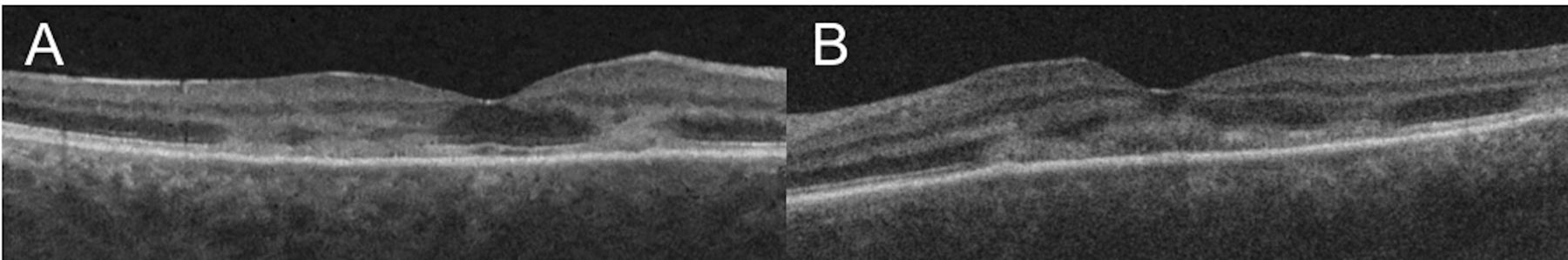
Clinical examination showed bilateral anterior uveitis, vitritis and multiple yellow-white placoid lesions at the level of the retinal pigment epithelium (RPE) at the posterior pole in both eyes (figure 1A,B). Fundus fluorescein angiography (FFA) showed correlating well-circumscribed lesions with early dense hypofluoroescence, followed by late hyperfluoroescence (figure 1C,D). Optical coherence tomography (OCT) showed hyperreflectivity and disruption to the outer retinal layers from outer plexiform layer (OPL) to the RPE in both foveae (figure 2A,B). Serological investigations and a chest X-ray ruled out infectious, autoimmune and inflammatory aetiologies while MRI of the brain ruled out central nervous system (CNS) vasculitis. Ocular differential diagnoses included infectious uveitis (syphilis, tuberculosis, viral, fungal, toxoplasma), choroidal metastases and ‘white-dot syndromes’ such as serpiginous chorioretinitis, multiple evanescent white dot syndrome, birdshot chorioretinopathy, Vogt-Koyanagi-Harada syndrome and multifocal choroiditis. Neurological differentials included lymphoma, autoimmune-related vasculitis, sarcoidosis and meningitis.

(A, B) Widefield fundus photograph (Optos ‘California’, Optos, Scotland) of the right and left eye, respectively, showing multiple creamy yellow-white placoid lesions at the posterior pole. (C, D) widefield late phase fluorescein angiogram of the right and left eye, respectively, showing dense hypofluorescent placoid lesions surrounded by late irregular hyperfluorescent leakage.
{kind=link}
{kind=link}
(A, B) Macular optical coherence tomography (Cirrus 5000, Carl Zeiss Meditec, Dublin, USA) of the right and left eye, respectively, showing hyperreflectivity from to the outer retinal layers from outer plexiform layer to the RPE with associated disruption and loss of the ellipsoid zone in both foveae. RPE, retinal pigment epithelium.
Given the correlation of symptoms and signs, and the absence of another diagnosis, a diagnosis of acute posterior multifocal placoid pigment epitheliopathy (APMPPE) was made and the patient was started on intravenous methylprednisolone therapy for 3 days, followed by oral prednisolone. After steroid therapy, the patient noted a slow return of BCVA to 6/9 OU with improvement in systemic symptoms.
APMPPE is a bilateral idiopathic inflammatory chorioretinopathy classified as a white dot syndrome.1 APMPPE has an estimated incidence of 0.15 cases per 100 000 persons with a peak onset between the second to fourth decades and no gender predilection.2 3 Risk factors for APMPPE include autoimmune diseases,3 infection4 and human leucocyte antigens B7/DR2 haplotypes.5 Interestingly, some reports have described the onset of APMPPE after vaccinations for polio, tetanus, varicella, hepatitis A/B, meningococcal C, yellow fever, typhoid, influenza and COVID-19.4 6 7
Patients with APMPPE can report asymmetric, subacute visual impairment with central/paracentral scotoma, photopsia and/or metamorphopsia.8 One-third of APMPPE cases experience a viral prodrome prior to APMPPE symptom onset.1 9 Neurological symptoms such as headache and sensorineural hearing loss may require imaging to rule out CNS vasculitis.10
Hallmark clinical features include bilateral, multiple, large (1–2 disc diameters), creamy yellow-white placoid lesions at the level of RPE and choroid throughout the posterior pole.8 Associated findings may include mild anterior uveitis/vitritis/papillitis with cystoid macular oedema rare. FFA demonstrates early hypofluorescent lesions followed by late, irregular hyperfluorescence.8 11 OCT shows hyperreflective lesions from the OPL to the RPE with ellipsoid zone loss.12 13 When quiescent, focal photoreceptor/RPE atrophy may occur at site of resolved placoid lesions.9 12–14
APMPEE is usually self-limiting with resolution of visual symptoms typically occurring by 4–8 weeks. However, 25% of individuals may have foveal involvement and a visual prognosis limited to BCVA of ≥6/15.15 There is currently no consensus on treatment, however, steroids have been reported to be beneficial in cases with macular involvement or associated CNS vasculitis.12 APMPPE recurrence/persistent is rare and may represent relentless placoid chorioretinitis if lasting ≥6 months.2 16 17
Patient’s perspective
At the time, I was completely exhausted all the time and did not feel myself. When my vision became blurred I knew I had to see my general practitioner. Thankfully, after a number of weeks, I am feeling much better with much more energy.
Learning points
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) may develop as an immune response to vaccine administration and should be considered in young to middle-aged patients with bilateral vision impairment following SARS-CoV-2 mRNA vaccination.
Steroid treatment may be considered in APMPPE with foveal involvement to prevent visual acuity loss.
Patients with a new diagnosis of APMPPE and neurological symptoms may require a full neurological and systemic work up rule out potentially fatal central nervous system vasculitis.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors KM: responsible for manuscript creation. RM: responsible for learning points and overall editing of manuscript. EA: responsible for creation of figures and legends. EO’C: treating Ophthalmologist, provided oversight and guidance of project as well as consultant editing of manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.