
Abstract
Introduction
The monoclonal antibody therapies bamlanivimab (BAM) + etesevimab (ETE) received emergency use authorization (EUA) from the US Food and Drug Administration (February 9, 2021) for treatment of mild-to-moderate COVID-19. The EUA of BAM + ETE was revoked (December 14, 2023) due to the high prevalence of BAM + ETE-resistant variants of SARS-CoV-2. Efficacy and safety of 700/1400 mg and 2800/2800 mg BAM + ETE are well established and published; however, efficacy and safety of 350/700 mg BAM + ETE have not been disclosed to date.
Methods
This portion of phase 3, BLAZE-1 trial (J2X-MC-PYAB) enrolled patients (between June 17, 2020 and April 9, 2021) with mild-to-moderate COVID-19 within 3 days of laboratory diagnosis of SARS-CoV-2 infection. In total, 354 patients with at least one risk factor for severe COVID-19 were enrolled, randomized (2:3), and infused with placebo (N = 141) or 350/700 mg BAM + ETE (N = 213), over ~ 8 min. Primary endpoint was to assess proportion of patients with persistently high SARS-CoV-2 viral load (PHVL) (log viral load > 5.27) 7 days after infusion.
Results
Patients were aged (mean) 53 years, 49.7% female, and 82.7% White. Seven days after drug infusion, 10.8% (95% confidence interval: 6.6, 15.0; p < 0.001) of BAM + ETE-treated patients and 34.8% (26.9, 42.6) of placebo-treated patients had PHVL, and the viral load change from baseline (least square mean [standard error]) was − 3.50 (0.15; p < 0.001) in BAM + ETE-treated patients versus − 2.51 (0.19) in placebo-treated patients. The majority of treatment-emergent adverse events were considered mild or moderate in severity (BAM + ETE: 6.6%; placebo: 14.2%). No deaths were reported.
Conclusions
Consistent with previous studies, patients treated with BAM + ETE (350/700 mg) had a significantly lower proportion of PHVL and greater reduction in viral load compared with placebo. The overall safety profile is consistent with higher doses of BAM + ETE. Infusions of over ~ 8 min did not result in meaningful increase in incidence of TEAEs compared to higher doses of BAM + ETE administered over 30–60 min.
Trial Registration
Clinical trial.gov identifier, NCT04427501.
Similar content being viewed by others


Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Why carry out the study? |
During the COVID-19 pandemic, the limited supply of monoclonal antibody therapies was a notable challenge and impacted its distribution and access in individuals at high risk of progression to severe COVID-19. |
BLAZE-1 study assessed the efficacy and safety of lower doses and faster infusions of bamlanivimab (BAM) + etesevimab (ETE), monoclonal antibody therapies for COVID-19. The goal was to address the constrained supply and reduce the healthcare burden. |
What was learned from the study? |
Results from BLAZE-1 study demonstrated that BAM + ETE therapy resulted in a significantly lower proportion of persistently high SARS-CoV-2 viral load and significantly greater reduction in viral load in the BAM + ETE group compared to the placebo. |
BLAZE-1 results demonstrated that the safety and efficacy of low-dose BAM + ETE was similar to the previously authorized high dose of BAM + ETE in patients with COVID-19. |
Introduction
Monoclonal antibody (mAb) therapies have been recommended by the National Institutes of Health (NIH) for the ambulatory treatment or prevention of COVID-19 in at-risk patients [1]. Neutralizing mAbs such as bamlanivimab (BAM) and etesevimab (ETE) inhibit SARS-CoV-2 viral attachment and entry into target cells by binding the receptor binding domain (RBD) of the spike protein of SARS-CoV-2 [2,3,4,5]. BAM and ETE together received Emergency Use Authorization (EUA) in February 2021, for the treatment of mild-to-moderate COVID-19 and for post-exposure prophylaxis of COVID-19 in at-risk adults and pediatric patients (12 years of age or older). The EUA for BAM + ETE for the treatment or prevention of COVID-19 was later revoked on December 14, 2023 [7], which was not due to any new safety concerns. BAM + ETE were designed to be effective against the early strains of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Authorization was based on the 700/1400 mg BAM + ETE combination [7], but the efficacy, safety, and benefits of lower BAM + ETE combinations have yet to be disclosed.
EUA was supported by clinical results from the phase 2/3 randomized and placebo-controlled BLAZE-1 (PYAB; NCT04427501) trial demonstrating that BAM + ETE can reduce COVID-19-related hospitalizations and deaths, increase overall viral clearance, and improve the time to COVID-19 symptom resolution. By day 29, 2.1% (11/518) patients at increased risk for severe COVID-19 treated with BAM + ETE had a COVID-19-related hospitalization or death from any cause, compared with 7.0% (36/517) patients who received placebo (absolute risk difference, – 4.8%; 95% confidence interval [CI], – 7.4 to – 2.3; relative risk difference, 70%, p < 0.001) [8]. No deaths were reported in the patients treated with BAM + ETE; ten deaths were reported in the patients treated with placebo, of which nine deaths were COVID-19–related as designated by the trial investigators [8]. Positive clinical outcomes with BAM + ETE were associated with rapid viral clearance as well as the absence of persistently high viral load (PHVL) 7 days following initiation of treatment [9].
During the initial stages of the pandemic, the limited supply of mAb therapies remained a notable challenge and impacted the distribution of mAb therapies to high-risk patients in need of therapy [10], warranting further investigation to confirm the efficacy of lower doses of BAM + ETE to increase the available supply for at-risk patients. Results from the BLAZE-4 phase 2 study (NCT04634409) suggested improved viral clearance with a low dose of 350/700 mg BAM + ETE relative to placebo in patients without risk factors for severe disease (i.e., age > 65 years and/or obesity) [11]. Therefore, later BLAZE-1 trial arms were designed to test the safety and efficacy of lower-dose 350/700 mg BAM + ETE in patients with increased risk for severe disease; further, the safety of faster infusions was investigated, with the ultimate goal being to increase access and decrease healthcare burdens. In these subsequent trial arms, BAM + ETE could be administered via intravenous (IV) push or syringe pump. IV push delivers a single dose of medicine directly into the bloodstream within a short period of time [12].
A total of 354 patients were randomized and infused with either low-dose BAM + ETE (N = 213) or placebo (N = 141) within 3 days of a positive SARS-CoV-2 test (BLAZE-1, NCT04427501). Patients were infused over approximately 8 min with either an IV push or a syringe pump. The primary objective was to assess the proportion of patients with PHVL 7 days after drug infusion. Patients with ribonuclease P normalized viral load > 5.27 were considered to have PHVL. The safety of low-dose BAM + ETE rapid IV push infusion at 3 and 5 min was also investigated. This manuscript presents the previously undisclosed effects of the safety and efficacy of low-dose 350 mg BAM and 700 mg ETE administered together and after rapid IV push infusion.
Methods
Study Design
A total of 354 ambulatory patients from the U.S., aged ≥ 12 years were evaluated in this portion of the phase 3, BLAZE-1 trial (J2X-MC-PYAB). The primary results of BLAZE-1 have been reported previously [13]. The primary objective of the study was to evaluate PHVL (PHVL was defined as a log viral load > 5.27, corresponding to a mean PCR cycle-threshold value of < 27.5 on day 7) in patients treated with low dose (350/700 mg) BAM + ETE compared to placebo. Secondary objectives included effects on SARS-CoV-2 viral load, clinical status, and symptom resolution in BAM + ETE and placebo groups. The rationale for the BAM + ETE dose selection was partly based on positive results in the BLAZE-4 study [11] supporting the potential efficacy of low-dose BAM + ETE for mild-to-moderate COVID-19 in patients without risk factors for severe disease (i.e., age > 65 years and/or obesity). Patients were infused with a total dose volume of 200 ml over at least 8 min and monitored for at least 60 min after completion of the infusion. Patients assigned to placebo treatment received 200 ml 0.9% sodium chloride injection. Each participant received a single dose of study intervention, and patients were followed for 85 days post-treatment.
Separately, a total of 300 ambulatory patients ≥ 12 years of age were treated with 5- or 3-min IV push of low-dose (350/700 mg) BAM + ETE in an addendum to the BLAZE-1 study. The primary objective was to evaluate the safety and clinical status after a single dose of BAM + ETE IV push during an 85-day post-treatment follow-up.
Eligibility
Inclusion and exclusion criteria are consistent with other reported results of the BLAZE-1 study [8]. In brief, at the time of screening, patients were ≥ 12 years of age and satisfied at least one high-risk criterion had one or more mild or moderate COVID-19 symptoms based on FDA guidance [14], were not currently hospitalized, and had a positive SARS-CoV-2 PCR test ≤ 3 days prior to the start of drug infusion. For ethical reasons, once the clinical efficacy of BAM + ETE at higher doses was established, enrollment into the placebo arm was stopped in this trial to ensure that patients were not deprived of potential treatment benefits.
The trial complied with the Declaration of Helsinki, the International Conference on Harmonization Guidelines for Good Clinical Practice, and applicable local regulations. The protocol was reviewed and approved by the ethics committees of all participating centers, and all patients or legally authorized representatives gave written informed consent prior to study entry.
Primary Endpoint: PHVL
PHVL was defined as a viral load greater than 5.27 on day 7 as validated by Dougan et al. [9]. The proportion of patients with PHVL was summarized and compared to placebo.
Secondary Endpoints: Viral Load, Clinical Status, Symptom Improvement and Resolution, and Safety
Viral load change from baseline (CFB) was calculated as the value at the visit of interest minus the baseline value. The baseline was defined as the last non-missing assessment recorded on or prior to the date of study drug infusion. Viral RNA was collected from nasopharyngeal swabs of patients. The proportion of patients experiencing COVID-19 hospitalization (defined as ≥ 24 h of acute care) or death from any cause by day 29 was compared for the treatment arm to all high-risk placebo patients. To increase the power of the analysis of this outcome measure, patients from the previous phase 3 BLAZE-1 placebo arm (N = 776) (these patients were from the corresponding placebo group for the phase 3 high-risk population, who were administered BAM + ETE 2800 mg + 2800 mg and the BAM + ETE 700 mg + 1400 mg) were pooled with the concurrent placebo group (N = 141).
Symptom improvement and resolution were assessed with symptom questionnaires completed by the patients. Patients rated the severity of their COVID-19 symptoms daily through day 11, and on day 22 and day 29, on a scale of none/absent = 0; mild = 1; moderate = 2; and severe = 3. The time to symptom improvement and resolution were summarized using Kaplan–Meier estimates through day 29. The time to symptom improvement was defined as the time to a patient experiencing both any symptoms on the symptom questionnaire scored as moderate or severe at baseline are subsequently scored as mild or absent, and any symptoms on the symptom questionnaire scored as mild or absent at baseline are subsequently scored as absent. The time to sustained symptom resolution was defined as the time to two consecutive assessments with a score of 0 for shortness of breath, fever, body aches, sore throat, chills, and headaches, or a score of 0 or 1 for cough and fatigue on the symptom questionnaire. The time to sustained complete symptom resolution was defined as the time to the first of two consecutive days that all symptoms had been given a score of 0.
The proportion of patients experiencing treatment-emergent adverse events (TEAEs), defined as an event that first occurred or worsened in severity after baseline, was also summarized. COVID-19 signs and symptoms were not considered TEAEs in the study.
Statistical Analysis
Patients were randomized 2:3 (placebo: 350/700 mg). A sample size of approximately 400 patients provides greater than 90% power to demonstrate that the effectiveness of BAM + ETE is statistically significantly better than placebo. This sample size calculation assumed a placebo event rate of 30% and a relative reduction of 60% for BAM + ETE, which were included in available data on PHVL rates. Because enrollment was stopped to ensure that patients were not randomized to placebo once the clinical efficacy of BAM + ETE was established, the study was not fully enrolled as originally planned.
The efficacy population included patients from the safety population who also had at least one non-missing post-baseline measurement. For continuous measures, the number of patients, mean, standard deviation, median, minimum, and maximum were included. For categorical measures, frequency counts and percentages were included.
Treatment comparisons for the proportion of patients with PHVL on day 7 were conducted using a logistic regression with a Firth penalized likelihood [15]. Missing viral load data on day 7 were imputed using the last observation carried forward (LOCF).
Change from baseline to days 3, 5, 7, and 11 in SARS-CoV-2 viral load was analyzed using a linear mixed-effect model. The model included log base 10 transformed baseline as a covariate, treatment, day, and treatment-by-day interaction as fixed effects. Missing viral load data were treated as missing at random.
The proportion of patients experiencing a COVID-19-related hospitalization or death from any cause was analyzed using logistic regression with a Firth penalized likelihood for the safety population. The model included a covariate for the duration from symptom onset to randomization (≤ 8 days, or > 8 days). Missing data were not imputed.
Time to symptom improvement, time to sustained, sustained complete, and complete symptom resolution were analyzed using a log-rank test stratified by duration from symptom onset to randomization. Patients who were either hospitalized, discontinued early, or did not experience an event by study completion were censored. Safety data were summarized descriptively. The final database lock occurred on October 10, 2021. The safety population included patients randomly assigned to treatment and who received any amount of study intervention.
Results
Patients were evaluated for eligibility and randomized to receive 350/700 BAM + ETE (N = 213) or placebo (N = 141) (Fig. 1). Patients were aged 53 years (mean), 82.7% were White, 12.5% were Black or African American, and 25.2% were Hispanic. The majority (81.9%) had mild COVID-19 and 97.7% of patients were at high risk of severe COVID-19 (Table 1). A few (4.0% [14/354]) patients had received the COVID-19 vaccine prior to the baseline assessment.

Participant enrollment. BAM bamlanivimab, ETE etesevimab, PHVL persistently high SARS-CoV-2 viral load
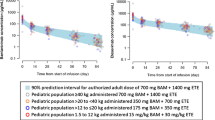
A significantly lower proportion of patients treated with BAM + ETE experienced PHVL at day 7 compared to placebo-treated patients, with an overall significant reduction in PHVL of – 24.0 (95% CI, − 32.9 to − 15.1, p < 0.001). The day 7 PHVL incidence was significantly lower in the BAM + ETE group (10.8%) versus the placebo group (34.8%) (Fig. 2). There was a significantly greater reduction (p < 0.001) in the viral load in the BAM + ETE versus the placebo group from baseline to days 3 (− 1.53 vs. − 0.75), 5 (− 2.83 vs. − 1.49), 7 (− 3.50 vs. − 2.51), and 11 (− 4.40 vs. − 3.79).

Effect of low-dose BAM + ETE on A PHVL at day 7 and B viral load- change from baseline. *Significantly greater reduction in the PHVL in the BAM + ETE group versus the placebo group at day 7 (p < 0.001). Values for PHVL at day 7 are presented as n (%). Baseline viral load (mean), BAM + ETE: 6.37; placebo: 6.74. BAM bamlanivimab, ETE etesevimab, Pbo placebo, PHVL persistently high SARS-CoV-2 viral load
COVID-19-related hospitalizations within 29 days after drug infusion were reported in 2 (0.9%) patients randomized to BAM + ETE (n = 213) and 60 (6.5%) patients randomized to placebo (n = 917) (Fig. 3). COVID-19-related deterioration by day 29, including hospitalization or death, was reported in 2/213 (0.9%) patients randomized to BAM + ETE and 61/917 (6.7%) patients randomized to placebo.

Effect of low-dose BAM + ETE on COVID-19-related hospitalizations within 29 days after drug infusion. P value vs. placebo, p < 0.005. Note: Patients in the placebo group were a combination of the corresponding placebo group enrolled concurrently with the BAM + ETE 2800 mg + 2800 mg-treated group and the corresponding placebo group enrolled concurrently with the BAM + ETE 350 mg + 700 mg-treated group. BAM bamlanivimab, ETE etesevimab, Pbo placebo
The time to symptom improvement (events in percentage; median in days [95% CI]) (BAM + ETE: 84.5%, 7.0 days [6.0, 8.0]; placebo: 84.4%, 9.0 days [7.0, 11.0], p = 0.69) was not significantly different in patients that received BAM + ETE compared to placebo (Table 2). Time to sustained symptom resolution (BAM + ETE: 79.8%, 6.0 days [5.0, 8.0]; placebo: 68.8%, 9.0 days [8.0, 13.0], p < 0.05), sustained complete symptom resolution (BAM + ETE: 61.0%, 12.0 days [10.0, 22.0]; placebo: 48.2%, 25.0 days [22.0, –] p < 0.05), and complete symptom resolution (BAM + ETE: 76.5%, 9.0 days [7.0, 10.0]; placebo: 68.8%, 22.0 days [11.0, 23.0], p < 0.05) were significantly lower in patients that received BAM + ETE compared to placebo. BAM + ETE treatment reduced the median days to resolution by 3 days for sustained symptom resolution, by 13 days for sustained completed symptom resolution, and 13 days for complete symptom resolution compared to placebo.
One patient in BAM + ETE group and four patients in placebo group reported at least one SAE. The SAE reported in the BAM + ETE group was femur fracture; and the SAEs reported in the placebo group were bone contusion, concussion, hip fracture, cardiac arrest, myocardial infarction, chest pain, COVID-19 pneumonia, pleural effusion, and respiratory arrest. These SAEs were not considered related to the study drug by the investigators. There was a higher incidence of TEAEs in patients treated with placebo compared to BAM + ETE group (14.2 vs. 6.6%). The patients treated with BAM + ETE experienced mild (2.3%) and moderate (4.2%) TEAEs, while those treated with placebo experienced mild (6.4%), moderate (6.4%), and severe (1.4%). There were two reported discontinuations from the study due to AEs (a urinary tract infection and a hip fracture, both in the placebo group). No deaths from AEs were reported from either of the groups (Table 3). Immediate hypersensitivity reaction, defined as events that occurred on the day of study drug administration, was reported in one participant treated with BAM + ETE and in none treated with placebo. Non-immediate hypersensitivity reactions, defined as a hypersensitivity event occurring > 6 h after study drug infusion, were reported in three patients treated with placebo and no BAM + ETE-treated patients.
An addendum to the BLAZE-1 protocol investigated the rapid infusion of 350/700 mg BAM + ETE for 5 min (n = 30) or 3 min (n = 270). In total, 44/300 (14.7%) patients that received rapid infusion 350/700 mg BAM + ETE reported TEAEs (Supplementary material). Ten (3.3%) of the total TEAEs reported were severe. SAEs were reported by 11 patients (one patient in 350/700 mg BAM + ETE 5-min infusion; ten patients in 350/700 mg BAM + ETE 3-min infusion). Anemia was reported as an SAE in patients treated with 350/700 mg BAM + ETE 5-min infusion and cardiac failure congestive, supraventricular tachycardia, diabetic ketoacidosis, hyperglycemic hyperosmolar nonketotic syndrome, generalized edema, bile duct stone, bacterial pneumonia, abnormal blood creatinine, abnormal glomerular filtration rate, rheumatoid arthritis, chronic obstructive pulmonary disease, hypertension was reported as SAEs in patients treated with 350/700 mg BAM + ETE 3-min infusion. There were no discontinuations from the study or deaths due to AEs for the patients treated with BAM + ETE. Immediate hypersensitivity in 5-min infusion was reported in one patient. Anaphylactic reactions were reported in two patients in 5-min and two patients in 3-min infusions. Nonimmediate hypersensitivity was reported in one patient in 5-min infusion and one patient in 3-min infusion. Three patients reported anaphylactic shock in 3-min infusion.
Discussion
Low-dose BAM + ETE significantly reduced viral load and the proportion of patients with PHVL 7 days after infusion and reduced the proportion of patients with COVID-19-related hospitalization 29, 60, and 85 days after infusion compared to placebo. Low-dose BAM + ETE also improved symptom resolution under several definitions compared to placebo. The safety profile of the lower dose of BAM + ETE is consistent with higher doses [13] with no new safety concerns.
The difference in PHVL at day 7 after low-dose BAM + ETE compared to placebo was − 24.0% (− 32.9 to − 15.1) and was similar to the difference in PHVL at day 7 observed with the authorized dose of 700/1400 BAM + ETE [9] (∆− 26.2, − 32.9 to − 19.4) and after 2800/2800 mg BAM + ETE [8] dose reported (∆19.6, − 24.4 to − 14.9). PHVL at day 7 was identified as a feature predictive of progression to severe COVID-19; amongst patients who were hospitalized with COVID-19 or had died from any cause by day 29 in earlier treatment arms of the phase 2/3 BLAZE-1 trial, 69% (45 of 65 patients) of the phase 3 population (composed solely of high-risk patients) had PHVL at day 7. In patients who were never hospitalized, only 23% (403 of 1733 patients) had PHVL on day 7, respectively [9].
Viral load change from baseline at 7 days was also similar after low-dose BAM + ETE compared to the 700/1400 mg and 2800/2800 mg BAM + ETE [8, 9]. The low-dose BAM + ETE change from baseline at 7 days (LSM [SE]) was − 3.50 (0.15) compared to − 2.51 (0.19) for placebo, resulting in a difference of − 0.99 (− 1.45 to − 0.52) for low-dose BAM + ETE compared to − 0.99 − 1.33 to − 0.66) for the authorized 700/1400 mg BAM + ETE dose and − 1.20 (− 1.46 to − 0.94) for the 2800/2800 mg BAM + ETE dose. These similarities among different doses and patient groups suggest that the low-dose BAM + ETE is similarly efficacious to the 700/1400 mg and 2800/2800 mg BAM + ETE doses in reducing the viral load.
The number of COVID-19-related hospitalizations from any cause across BAM + ETE treatments was also similar. There were 2/213 (0.9%) hospitalizations within 29 days in patients randomized to BAM + ETE, compared to 60/917 (6.5%) in pooled placebo. Similarly, there were 4/511 (0.8%) hospitalizations within 29 days after 700/1400 mg BAM + ETE compared to 15/258 (5.8%) after placebo [9], and 11/518 (2.1%) hospitalizations within 29 days after 2800/2800 mg BAM + ETE compared to 36/517 (7.0%) after placebo [8]. These data reveal consistent and reproducible reductions in COVID-19-related hospitalizations from any cause across BAM + ETE doses.
An addendum to BLAZE-1, PYAB additionally incorporated 5- and 3-min IV push treatments of low-dose BAM + ETE in order to support IV administration over a shorter time period. The ultimate goals of shorter treatment times were to reduce contact time with overburdened healthcare facilities and improve participant convenience and compliance. Compared to the standard infusion, a higher incidence of TEAEs (14.7% compared to 9.6%) and SAEs (3.7% compared to 1.4%) was observed with the shorter 5- and 3-min IV push treatments but similar frequencies of immediate hypersensitivity (0.3% for both), and non-immediate hypersensitivity (0.7% compared to 0.8%) were reported. On the other hand, the overall TEAEs and SAEs with the standard infusion (which was < 8 min infusion) of lower-dose BAM + ETE (350 mg + 700 mg) were consistent with the higher dose of BAM + ETE (2800 mg + 2800 mg) as reported in the primary BLAZE-1 study results [13]. The increased risk of AEs with the shorter 5- and 3-min IV push indicates that maintaining the standard infusion duration optimally ensured patient safety and minimized potential complications.
BAM + ETE is effective at neutralizing Delta (B.1.617.2/AY.3) and Alpha (B.1.1.7) variants, but has reduced efficacy against Omicron variants (B.1.1.529/BA.1) [7, 16, 17]. The emergence of Omicron as the predominant SARS-CoV-2 variant in the United States resulted in the revocation of the EUA of BAM + ETE.
A current limitation of the reported results includes a lack of baseline and treatment-emergent variant analyses that could facilitate a further understanding of the specific efficacy of low-dose BAM + ETE treatment. Also, the study was conducted when the original Wuhan strain and the subsequent Alpha variant were predominant, so the outcome from this study would not be generalizable in other patient populations with different strains of COVID-19.
Placebo patients from a previous phase 3, Blaze-1 study were included with placebo patients within this study during analysis of clinical outcomes (Fig. 3). Although placebo groups were enrolled within a few weeks of each other, the non-concurrent enrollment and infusion dates are a limitation of the study. Additional patients who received placebo, were added based on statistical and ethical considerations, such that studies were supported with higher power for statistical measures of low-frequency clinical outcomes (i.e., COVID-19-related deterioration and death) and more patients received the beneficial active treatment.
Overall, the efficacy and safety profiles of low-dose BAM + ETE at either standard or accelerated infusion rates were similar to those observed with the previously published and/or authorized doses of 700/1400 mg BAM + ETE and 2800/2800 mg BAM + ETE. Although efficacy results indicate the potential to lower the authorized BAM + ETE dose further below 700/1400, the time period during which the low-dose arms (January 20, 2021 to June 16, 2021) and rapid IV push arms of BLAZE-1 (March 11, 2021 to July 2, 2021) enrolled prevents a clear understanding of the impact of low-dose BAM + ETE on the dynamic variant distribution of COVID-19. The mounting concern of variant emergence and the unpredictability of the variant landscape thus prevented modifications to the authorized dose at 700/1400 BAM + ETE based on low-dose efficacy results.
Conclusions
This study demonstrated that treatment with BAM + ETE resulted in a significantly lower proportion of patients with PHVL compared to placebo-treated patients. Also, PHVL incidence was significantly lower and a significantly greater reduction in viral load was observed in the BAM + ETE group compared to placebo. Furthermore, COVID-19-related deterioration including hospitalization and death was overall lower in the BAM + ETE group indicating that the safety and efficacy of low-dose BAM + ETE was similar to the previously authorized high dose of BAM + ETE.
Data Availability
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date for data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data-sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms, will be provided in a secure data-sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
References
COVID-19 Treatment Guidelines. National Institute of Health. Available: https://www.covid19treatmentguidelines.nih.gov/therapies/anti-sars-cov-2-antibody-products/anti-sars-cov-2-monoclonal-antibodies/. Accessed 31 Mar 2022.
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–8.
Wang Q, Zhang Y, Wu L, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181(894–904):e9.
Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–4.
Benton DJ, Wrobel AG, Xu P, et al. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature. 2020;588:327–30.
Bamlanivimab/Etesevimab. 2023. Available https://aspr.hhs.gov/COVID-19/Therapeutics/Products/Bamlanivimab-etesevimab/Pages/default.aspx. Accessed 22 Apr 2024.
Fact sheet for health care providers Emergency Use Authorization (EUA) of bamlanivimab and etesevimab. Food and Drug Administration, 2021. Available: https://www.fda.gov/media/145802/download. Accessed 22 Feb 2022.
Dougan M, Nirula A, Azizad M, et al. Bamlanivimab plus etesevimab in mild or moderate Covid-19. N Engl J Med. 2021;385:1382–92.
Dougan M, Azizad M, Mocherla B, et al. A randomized, placebo-controlled clinical trial of bamlanivimab and etesevimab together in high-risk ambulatory patients with COVID-19 and validation of the prognostic value of persistently high viral load. Clin Infect Dis. 2021;75:e440–9.
Behr CL, Joynt Maddox KE, Meara E, Epstein AM, Orav EJ, Barnett ML. Anti-SARS-CoV-2 monoclonal antibody distribution to high-risk Medicare beneficiaries, 2020–2021. JAMA. 2022;327:980–3.
Nichols RM, Macpherson L, Patel DR, Yeh WW, Peppercorn A. Effect of bamlanivimab as monotherapy or in combination with etesevimab or sotrovimab on persistently high viral load in patients with mild-to-moderate COVID-19: a randomized, phase 2 BLAZE-4 trial. Infect Dis Ther. 2024;13(2):401–11.
IV PUSH VS IV INFUSION: WHICH ONE’S BETTER? 2017. Available: https://www.vitamininjections.co.uk/iv-push-vs-iv-infusion-which-is-better/. Accessed 7 Feb 2024.
Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA. 2021;325(7):632–44. https://doi.org/10.1001/jama.2021.0202
Guidance for industry. Covid-19: developing drugs and biological products for treatment or prevention. Food and Drug Administration, 2020. Available: https://www.fda.gov/media/137926/download. Accessed 3 Oct 2021.
Firth D. Bias reduction of maximum likelihood estimates Get access Arrow. Biometrika. 1993;80:27–38.
Sheward DJ, Kim C, Ehling RA, et al. Neutralisation sensitivity of the SARS-CoV-2 omicron (B.1.1.529) variant: a cross-sectional study. Lancet Infect Dis. 2022;22(6):813–20.
VanBlargan LA, Errico JM, Halfmann PJ, et al. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat Med. 2022;28(3):490–5.
Acknowledgements
We thank Karthik Chandrasekhar for assistance with statistical analysis.
Medical Writing Assistance
Writing and editorial assistance was provided by Adrija Tripathy and Divya Kamboj from Syneos Health and was funded by Eli Lilly and Company.
Funding
This study and the Rapid Service Fee for this publication were sponsored by Eli Lilly and Company.
Author information
Authors and Affiliations
Contributions
Dipak R. Patel, Lisa Macpherson, Martin Bohm, Himanshu Upadhyaya, Ajay Nirula, Paul Klekotka, Mark Williams, and Matthew M. Hufford have contributed to the acquisition of data for the work. Dipak R. Patel, Lisa Macpherson, Martin Bohm, Himanshu Upadhyaya, Carmen Deveau, Ajay Nirula, Paul Klekotka, Mark Williams, and Matthew M. Hufford have contributed to the interpretation of data for the work, drafting, and critical revision of the work for important intellectual content.
Corresponding author
Ethics declarations
Conflict of Interest
Dipak R. Patel, Lisa Macpherson, Martin Bohm, Carmen Deveau, Paul Klekotka, Mark Williams, and Matthew M. Hufford are employees and/or stockholders of Eli Lilly and Company. Himanshu Upadhyaya was an employee at Eli Lilly and Company at the time of study and development of the manuscript and is currently employed by Supernus Pharmaceuticals, Inc. Rockville, Maryland, USA. Ajay Nirula was an employee at Eli Lilly and Company at the time of study and development of the manuscript and is currently employed by Recludix Pharma, San Diego, California, USA.
Ethics Approval and Consent to Participate
The study was conducted in accordance with the principles of the Declaration of Helsinki (2000), the International Conference on Harmonization, and the E6 Guideline for Good Clinical Practice. Institutional review board approval and written informed consent from all subjects were obtained before the conduct of any evaluations or study procedures.
Consent for Publication
All authors have given their consent and approval of the final version to be submitted and published.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Patel, D.R., Macpherson, L., Bohm, M. et al. Efficacy and Safety of Low-Dose, Rapidly Infused Bamlanivimab and Etesevimab: Phase 3 BLAZE-1 Trial for Mild-to-Moderate COVID-19. Infect Dis Ther 13, 2123–2134 (2024). https://doi.org/10.1007/s40121-024-01031-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-024-01031-z