
Abstract
Advanced age correlates with higher morbidity and mortality among patients affected with the novel coronavirus disease 2019 (COVID-19). Because systemic inflammation and neurological symptoms are also common in severe COVID-19 cases, there is concern that COVID-19 may lead to neurodegenerative conditions such as Alzheimer’s disease (AD). In this review, we summarize possible mechanisms by which infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19, may cause AD in elderly COVID-19 patients and describe preventive measures to mitigate risk. Potential mechanisms include NLRP3 inflammasome activation and IL-1β release, renin-angiotensin system hyperactivation, innate immune activation, oxidative stress, direct viral infection, and direct cytolytic β-cell damage. Anti-inflammatory therapies, including TNF-α inhibitors and nonsteroidal anti-inflammatory drugs, antioxidants such as the vitamin E family, nutritional intervention, physical activity, blood glucose control, and vaccination are proposed as preventive measures to minimize AD risk in COVID-19 patients. Since several risk factors for AD may converge during severe SARS-CoV-2 infection, neurologists should be alert for potential symptoms of AD and actively implement preventive measures in patients presenting with neuropsychiatric symptoms and in high-risk patients such as the elderly.
Similar content being viewed by others
Introduction
About one year after its onset, the outbreak of the novel coronavirus disease 2019 (COVID-19) caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has evolved into an emergent global pandemic that impacted dramatically on the field of Alzheimer’s disease (AD) research [1]. There are several parallels between AD and COVID-19 in regard to pathogenic mechanisms and risk factors. Advanced age, for instance, is both the main risk factor for AD and the strongest predictor of COVID-19 mortality [2]. On the other hand, a broad range of neurological and neuropsychiatric symptoms, including loss of smell (anosmia) or taste (ageusia), headache, dizziness, and epilepsy, have been reported in patients infected with SARS-CoV-2 [3,4,5,6]. Since cognitive decline was also described in elderly COVID-19 patients, it was suggested that SARS-CoV-2 infection can lead to AD development and other long-term neurological sequelae [5, 7, 8]. Although direct evidence for COVID-19-induced AD has not so far surfaced, there are several possible mechanisms by which COVID-19 may initiate AD. These include systemic inflammation, renin-angiotensin system (RAS) hyperactivation, innate immune activation, oxidative stress, direct viral infection, and direct cytolytic β-cell damage. This review aims to systematically summarize hypothetical mechanisms of COVID-19-mediated AD onset and progression in elderly patients and puts forward some preventive measures to minimize those risks.
COVID-19 may cause cognitive dysfunction
Recent reports discussed evidence of neurodegeneration and cognitive impairment sequelae triggered by immunological issues in patients with neurological symptoms caused by COVID-19 [5, 9]. Indeed, an observational study in France reported that one-third of COVID-19 patients admitted due to acute respiratory distress syndrome (ARDS) had evidence of cognitive impairment at the time of discharge [10]. Therefore, it is particularly meaningful to summarize the potential mechanisms by which SARS-CoV-2 can cause AD.
Possible mechanisms of SARS-CoV-2-induced AD
Systemic inflammation
Mounting evidence suggests that neuroinflammation is involved in the pathophysiology of neurodegenerative diseases such as AD, a condition characterized by Aβ accumulation and tau phosphorylation [11,12,13]. The immunological sensor nucleotide-binding domain and leucine-rich repeat (NLR) pyrin domain-containing 3 (NLRP3) inflammasome is a key mediator of AD development [14, 15]. Studies have shown that the systemic inflammatory response induced by SARS-CoV-2 infection is mediated in part by overstimulation in the NLRP3 inflammasome pathway [16,17,18]. Critically, NLRP3 inflammasome activation can potentially aggravate or initiate AD by impairing microglial amyloid-beta (Aβ) peptide clearance [19]. Moreover, AD development in COVID-19 patients can be further aided by pro-inflammatory cytokines such as interleukin (IL)-1β, released upon NLRP3 inflammasome activation [14], or IL-17, IL-6, and tumor necrosis factor-α (TNF-α), produced by immune cells in response to the infection [20,21,22].
Activation of the NLRP3 inflammasome
The SARS coronavirus ORF3a protein was shown to induce the extrinsic apoptotic pathway in human cells [23] and to activate the NLRP3 inflammasome by promoting TNF receptor-associated factor 3 (TRAF3)-dependent ubiquitination of apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) [24]. On the other hand, ARDS, a common complication of severe COVID-19 resulting from dysregulated hyperinflammation, can rapidly stimulate the innate immune response and lead to NLRP3 inflammasome activation [25,26,27]. Therefore, SARS-CoV-2 infection can trigger activation of NLRP3 inflammasome through both direct (ORF3a) and indirect (ARDS) mechanisms. As we all know, ARDS patients needed the lung protective ventilatory strategies to reduce pulmonary morbidity, however, which almost always lead to hypercapnia. A study from Ding et al. reported that hypercapnia can strengthen the activation of NLRP3 inflammasome and enhance the release of pro-inflammatory IL-1β in the hypoxia-activated microglia [18]. Elevated IL-1β secretion via activating the NLRP3 can induce neuroinflammation, neuronal death and cognitive impairments, which might involve in the pathogenesis of AD. Additionally, these findings can be reinforced by this fact that apoptosis of neurons and impairments of cognitive function might be ameliorated though pharmacologically inhibiting NLRP3 inflammasome activation and IL-1β release. Therefore, SARS-CoV-2 infection might contribute to the pathogenesis of AD via activation of NLRP3 inflammasome and overproduction of IL-1β [18] (Fig. 1).

COVID-19 may cause AD by activating the NLRP3 inflammasome
Role of IL-17, IL-6, and TNF-α
Systemic inflammation caused by COVID-19 is characterized by the convergence of inflammatory mediators such as IL-17, IL-6, and TNF-α, among others [28]. IL-17, produced by T helper 17 cells, has been implicated in the pathogenesis of chronic inflammatory diseases such as AD [20]. The main targets of IL-17 are the neutrophils, which can promote inflammation and central nervous system (CNS) tissue damage upon stimulation by this cytokine. Through this mechanism, IL-17 plays an important role in AD pathology. TNF-α can link peripheral and central inflammation and was shown to modulate various neuropathological mechanisms in AD [29]. In turn, high levels of IL-6 in severe COVID-19 patients may predict hippocampal atrophy [22], which is one of the pathophysiological characteristics of AD.
Renin-angiotensin system (RAS) hyperactivation
There is evidence suggesting that angiotensin-converting enzyme 2 (ACE2) expression may be downregulated after binding of the receptor-binding domain of the S glycoprotein of SARS-CoV-2 to cellular ACE2 [28]. Downregulation of ACE2 may lead to increased expression of Angiotensin II (Ang-II) [28], the most important effector peptide of the RAS and a main systemic regulator of blood pressure [30]. In turn, SARS-CoV-2 may also stimulate RAS activity in the brain indirectly, by inducing the production of neurotoxins and proinflammatory factors acting on astrocytes [30]. Many tissues, such as the nigrostriatal system in the brain, are known to have a local RAS [30]. In the nigrostriatal system, RAS hyperactivation exacerbates oxidative stress and the microglial inflammatory response, contributing to dopaminergic degeneration and favoring AD development [30] (Fig. 2). Indeed, the contribution of the brain RAS to the development and progression of AD has been demonstrated by observational and experimental studies [31].

COVID-19 may cause AD via hyperactivation of the RAS
Innate immune activation
SARS-CoV-2 may enter the brain by binding to host cells, such as glial cells and neurons, that express the ACE2 receptor [32]. There is ample evidence that microglia, the brain’s major innate immune cells, play an indispensable role in AD pathogenesis by inducing neuroinflammatory responses that stimulate Aβ production [33]. Several key molecular and clinical manifestations of AD, such as Aβ accumulation, neuronal loss, and memory decline, might therefore be exacerbated after microglia become infected with SARS-CoV-2 [33].
Oxidative stress
Oxidative stress, triggered by a state of imbalance between reactive oxygen species (ROS) and the antioxidant defense system, has been reported to be involved in pathogenesis of SARS-CoV-2 infection [34]. This finding was further confirmed by a study by Panfoli et al. [35]. Interestingly, a study by Nasi et al. reported that production of ROS can induce lipid peroxidation [36]. Another research found that lipid peroxidation can cause oxidative dysfunction of key energy-related complexes in mitochondria and trigger neurodegeneration, which may eventually contribute to the development of AD [37]. This finding that SARS-CoV-2 may initiate AD by oxidative stress mechanism is consistent with the fact that oxidative stress, as we all know, is one of the pathogenesis of AD [22].
Direct viral infection
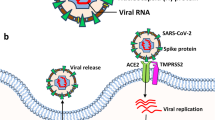
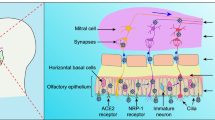
Like other human coronaviruses, SARS-CoV-2 has neuroinvasive, neurotropic, and neurovirulent properties [38]. Common neurological manifestations of SARS-CoV-2 infection include anosmia and ageusia, and clinical studies reported that olfactory and gustatory dysfunction affected a majority of patients with mild or severe COVID-19 [39, 40]. It is well known that some viral infections (e.g. the influenza virus) can cause loss of olfactory function [41]. Evidence suggests that SARS-CoV-2 can enter the CNS through the olfactory nerve in the nasal cavity by combination of its spike protein with ACE2 receptors in various cells of the olfactory epithelium, leading to loss of olfaction and eventually to viral dissemination to deeper structures in the brain [38]. It is reported that neurons and glial cells within brain structures such as the striatum, the substantia nigra, and brain stem express the ACE-2 receptor [28]. Upon entering the CNS, SARS-CoV-2 may directly activate innate immunity mediated by glial cells, potentially leading to Aβ accumulation and AD progression or onset [33]. Besides, direct infection of neurons within cognitive structures will cause neuroinflammation and neuronal necrosis, further stimulating the development of AD [18]. Importantly, reportedly elevated levels of ACE2 in AD patients may make them more susceptible to severe COVID-19 infection [42].
Direct cytolytic β-cell damage
There is an obvious relationship between AD and diabetes mellitus (DM) [43, 44]. It was estimated that the risk of developing AD is increased by 1.5- to twofold in long-term diabetic patients [43]. Glucose is the primary energy supply to the brain. By depriving the brain from insulin, DM decreases glucose metabolism, impairs cerebral blood supply, and disrupts normal cellular functions, promoting neurodegeneration and memory and cognitive deficits [44]. DM may also increase the aggregation of Aβ peptide, induce hyperphosphorylation of tau protein [44], and trigger deleterious changes in vascular structure and function [45]. Indeed, since hallmark molecular/clinical manifestations of AD can be triggered by insulin resistance in brain cells, AD has been referred to as “diabetes of the brain” or “type 3 diabetes” [46].
DM is considered a main risk factor for developing severe COVID-19 symptoms [47]. In turn, COVID-19 can exacerbate dysglycemia in people with DM, and evidence has emerged of diabetes-related symptoms, such as acute hyperglycemia and acute diabetic ketoacidosis, precipitated by SARS-CoV-2 in people with no history of diabetes [48]. It was thus proposed that SARS-CoV-2 can induce new-onset type 1 diabetes mellitus (T1DM) via direct cytolytic damage of pancreatic β-cells, which express the ACE2 receptor [48].
Potential preventive measures
As discussed in the above sections, the global spread of SARS-CoV-2 has had profound implications in the field of AD [1, 49,50,51]. Of particular concern is the evidence that the COVID-19-related death rate is higher for AD patients than for elderly COVID-19 cases without AD [52]. Because AD is incurable and there are few effective drug treatments to slow disease progression [53], it is urgently necessary to adopt preventive measures to avoid SARS-CoV-2 contagion in AD patients and to mitigate, especially in the elderly, the risk of developing AD following infection. The sections below summarize key preventive measures in such regard.
Anti-inflammatory therapy
Both severe COVID-19 and AD are characterized by systemic inflammation triggered by elevated levels of TNF-α and other pro-inflammatory factors. Therefore, anti-inflammatory therapies represent first-line interventions to mitigate risk of brain damage and to prevent AD initiation in COVID-19 patients. Several studies on mice, rats, and monkeys showed that treatment with TNF-α inhibitors can significantly reduce the burden of neurofibrillary tangles, amyloid precursor protein, and Aβ plaques [13]. Therefore, TNF-α inhibitors represent valuable prophylactic agents to prevent AD through immune modulation of the TNF inflammatory pathway. In addition, another study reported that long-term use of nonsteroidal anti-inflammatory drugs can prevent the occurrence of AD by blocking neuronal ectopic cell cycle events caused by microglial activation induced by Aβ oligomers [54].
Antioxidant therapy
Because oxidative stress is involved in the pathogenesis of both the COVID-19 and AD, antioxidant therapies can be useful to prevent the occurrence of AD in COVID-19 patients. Vitamin E and derivatives such as tocotrienol are powerful antioxidants that were shown to act in brain cells to mitigate oxidative stress and mitochondrial dysfunction, two inter-dependent phenomena closely associated with AD pathogenesis [55, 56]. In this regard, a recent study indicated that garcinoic acid, a natural vitamin E metabolite, can prevent Aβ oligomerization and deposition in the mouse brain [57].
Nutritional interventions
Mounting evidence suggests that the incidence and severity of AD can be significantly attenuated by modulation of the gut microbiota and cerebral Aβ production through dietary interventions [58]. The bioactive components of commonly consumed foods and dietary supplements include prebiotics, probiotics, and synbiotics. Among the prebiotics, wheat bran was shown to promote the growth of commensal bacteria, positively modulate the gut–brain axis, decrease neuroinflammation, and delay the occurrence and progression of AD [58, 59]. The role of dietary patterns and nutritional interventions in AD progression and prevention was recently reviewed by Rodriguez-Casado et al. [60].
Physical activity
Several risk factors related to AD, such as DM, hypertension, and heart disease [61] are modifiable through physical activity and moderate intensity exercise such as aerobic activities and balance and flexibility training [62, 63]. Main benefits of physical activity on AD prevention include anti-inflammatory and antioxidative effects, as well as increased cerebral blood flow [62].
Blood glucose control
Some pathophysiological factors shared by DM and AD, such as chronic inflammation, oxidative stress, and mitochondrial dysfunction can be directly linked to abnormal glucose homeostasis [64]. As discussed above, DM is a risk factor for AD [64]. It is thus possible that COVID-19 may aggravate AD symptoms or stimulate its development by inducing dysglycemia and even T1DM [48]. Therefore, blood glucose control strategies represent a wise preventive approach to minimize AD risk and pathology in individuals affected by COVID-19. Main consequences of unstable blood glucose levels include microvascular complications, atherosclerosis, and severe hypoglycemic events [65]. As reviewed by Fiore et al. both hyperglycemia and hypoglycemia can induce cognitive decline and AD [64]. The potential therapeutic benefit of successful stabilization of blood glucose levels on SARS-CoV-2-related AD progression or risk is highlighted by a study from Sardu et al. who reported that insulin infusion can effectively achieve glycemic targets and improve poor prognosis in patients with COVID-19 [66].
Anti-Aβ vaccine that has the potentially promising
Since available treatments for AD show at best modest therapeutic effects, researchers have undertaken intensive efforts to develop vaccines able to halt AD progression by removing Aβ from the brain [67]. However, following promising results in mice, a clinical trial examining the effectiveness of vaccination with full length Aβ42 was stopped after 6% of patients with mild to moderate AD developed meningoencephalitis [68]. Follow-up studies showed however both partial and extensive plaque clearance in post-mortem AD brains from some vaccinated patients, although this did not translate into improved cognitive and disability scores [69]. This experience paved the way to further development of vaccine formulations targeting more specific disease epitopes and offering better control of the immune response [68]. In this regard, a recent study in the APP/PS1 mouse model of AD showed that injection of dendritic cells presenting a modified Aβ peptide triggered a specific antibody response and improved memory performance to a similar degree than that exhibited by non-transgenic, untreated mice. Importantly, the vaccine did not trigger an inflammatory response [70]. Despite past failures and the significant challenge involved in developing and testing vaccines against endogenous neurodegenerative disease proteins, these sustained efforts to harness the immune system to treat AD will hopefully bring long-awaited success.
Conclusions
The ongoing COVID-19 pandemic is having a major impact in both AD patients and AD research. CNS symptoms such as anosmia, ageusia, headache, dizziness, epilepsy, and cognitive decline, as well as systemic inflammation, RAS activation, innate immune activation, oxidative stress, direct viral infection, and direct cytolytic β-cell damage may all contribute to aggravate or initiate AD in people affected by COVID-19. Accordingly, preventive measures in the form of anti-inflammatory and antioxidant therapies appear particularly necessary to mitigate AD onset and progression caused by SARS-CoV-2. Further measures, including nutritional interventions, increased physical activity, and glycemic control can further help prevent the occurrence of AD in people at risk for developing severe COVID-19 symptoms. Although there are currently few studies addressing the relationship between COVID-19 and AD, the numerous potential links between the two diseases reinforce the need for comprehensive assessment of neurological symptoms and implementation of prophylactic and preventive measures to minimize AD risk following infection with SARS-CoV-2.
Data Availability
Data and materials can be made available upon request.
Code Availability
Not applicable.
Change history
20 October 2021
A Correction to this paper has been published: https://doi.org/10.1007/s10072-021-05680-7
References
Naughton SX, Raval U, Pasinetti GM (2020) Potential novel role of COVID-19 in Alzheimer’s disease and preventative mitigation strategies. J Alzheimers Dis 76(1):21–25. https://doi.org/10.3233/JAD-200537
Blagosklonny MV (2020) From causes of aging to death from COVID-19. Aging Albany NY 12(11):10004–10021. https://doi.org/10.18632/aging.103493
Beach S, Praschan N, Hogan C, Dotson S, Merideth F, Kontos N, Fricchione G, Smith F (2020) Delirium in COVID-19: a case series and exploration of potential mechanisms for central nervous system involvement. Gen Hosp Psychiatry 65:47–53. https://doi.org/10.1016/j.genhosppsych.2020.05.008
Niazkar H, Zibaee B, Nasimi A, Bahri N (2020) The neurological manifestations of COVID-19: a review article. Neurol Sci 41(7):1667–1671. https://doi.org/10.1016/10.1007/s10072-020-04486-3
Heneka MT, Golenbock D, Latz E, Morgan D, Brown R (2020) Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimers Res Ther 12(1):69. https://doi.org/10.1186/s13195-020-00640-3
Zhang J, Wang HL, Wei M, Zhang HZ, Xia BM, Wang XD, Pei YL, Dong L, Li YP (2020) Incidence of cerebrovascular disease as a comorbidity in patients with COVID-19: a meta-analysis. Aging (Albany NY) 12(23):23450–23463. https://doi.org/10.18632/aging.104086
Mohammadi S, Moosaie F, Aarabi M (2020) Understanding the immunologic characteristics of neurologic manifestations of SARS-CoV-2 and potential immunological mechanisms. Mol Neurobiol 57(12):5263–5275. https://doi.org/10.1007/s12035-020-02094-y
Wang FZ, Kream RM, Stefano GB (2020) Long-term respiratory and neurological sequelae of COVID-19. Med Sci Monit 26: e928996. https://doi.org/10.12659/MSM.928996
Alonso-Lana S, Marquié M, Ruiz A, Boada M (2020) Cognitive and neuropsychiatric manifestations of COVID-19 and effects on elderly individuals with dementia. Front Aging Neurosci 12:588872. https://doi.org/10.3389/fnagi.2020.588872
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, Collange O, Boulay C, Fafi-Kremer S, Ohana M, Anheim M, Meziani F (2020) Neurologic features in severe SARS-CoV-2 infection. N Engl J Med 382(23):2268–2270. https://doi.org/10.1056/NEJMc2008597
Moradi Majd R, Mayeli M, Rahmani F (2020) Pathogenesis and promising therapeutics of Alzheimer disease through eIF2alpha pathway and correspondent kinases. Metab Brain Dis 35(8). https://doi.org/10.1007/s11011-020-00600-8
Valmikinathan K, Verghese NJJocp (1966) Simple colour reaction for alkaptonuria. J Clin Pathol 19(2):200. https://doi.org/10.1136/jcp.19.2.200
Shamim D, Laskowski M (2017) Inhibition of inflammation mediated through the tumor necrosis factor α biochemical pathway can lead to favorable outcomes in Alzheimer disease. J Cent Nerv Syst Dis 9:117957351772251. https://doi.org/10.1177/1179573517722512
McKee C, Coll R (2020) NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. J Leukoc Biol 108(3):1–16. https://doi.org/10.1002/JLB.3MR0720-513R
Pennisi M, Crupi R, Di Paola R, Ontario M, Bella R, Calabrese E, Crea R, Cuzzocrea S, Calabrese V (2017) Inflammasomes, hormesis, and antioxidants in neuroinflammation: role of NRLP3 in Alzheimer disease. J Neurosci Res 95(7):1360–1372. https://doi.org/10.1002/jnr.23986
Polidoro R, Hagan R, de Santis SR, Schmidt N (2020) Overview: systemic inflammatory response derived from lung injury caused by SARS-CoV-2 infection explains severe outcomes in COVID-19. Front Immunol 11:1626. https://doi.org/10.3389/fimmu.2020.01626
Bouayad A (2020) Innate immune evasion by SARS-CoV-2: comparison with SARS-CoV. Rev Med Virol 30(6):e2135. https://doi.org/10.1002/rmv.2135
Ding HG, Deng YY, Yang RQ, Wang QS, Jiang WQ, Han YL, Huang LQ, Wen MY, Zhong WH, Li XS, Yang F, Zeng HK (2018) Hypercapnia induces IL-1beta overproduction via activation of NLRP3 inflammasome: implication in cognitive impairment in hypoxemic adult rats. J Neuroinflammation 15(1):4. https://doi.org/10.1186/s12974-017-1051-y
Tejera D, Mercan D, Sanchez-Caro JM, Hanan M, Greenberg D, Soreq H, Latz E, Golenbock D, Heneka MT (2019) Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. Embo J 38(17):e101064. https://doi.org/10.15252/embj.2018101064
Milovanovic J, Arsenijevic A, Stojanovic B, Kanjevac T, Arsenijevic D, Radosavljevic G, Milovanovic M (2020) Arsenijevic N (2020) Interleukin-17 in chronic inflammatory neurological diseases. Front Immunol 11:947. https://doi.org/10.3389/fimmu.2020.00947
Kalovyrna N, Apokotou O, Boulekou S, Paouri E, Boutou A, Georgopoulos S (2020) A 3’UTR modification of the TNF-α mouse gene increases peripheral TNF-α and modulates the Alzheimer-like phenotype in 5XFAD mice. Sci Rep 10(1):8670. https://doi.org/10.1038/s41598-020-65378-2
Munoz P, Ardiles AO, Perez-Espinosa B, Nunez-Espinosa C, Paula-Lima A, Gonzalez-Billault C, Espinosa-Parrilla Y (2020) Redox modifications in synaptic components as biomarkers of cognitive status, in brain aging and disease. Mech Ageing Dev 189:111250. https://doi.org/10.1016/j.mad.2020.111250
Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, Han Y, Zhang XY, Zhou W, Qiu Y, Zhou X (2020) The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol 17(8):881–883. https://doi.org/10.1038/s41423-020-0485-9
Siu KL, Yuen KS, Castano-Rodriguez C, Ye ZW, Yeung ML, Fung SY, Yuan S, Chan CP, Yuen KY, Enjuanes L, Jin DY (2019) Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J 33(8):8865–8877. https://doi.org/10.1096/fj.201802418R
Freeman TL, Swartz TH (2020) Targeting the NLRP3 inflammasome in severe COVID-19. J Med Virol 11:1518. https://doi.org/10.3389/fimmu.2020.01518
Yaqinuddin A, Kashir J (2020) Novel therapeutic targets for SARS-CoV-2-induced acute lung injury: targeting a potential IL-1β/neutrophil extracellular traps feedback loop. Med Hypotheses 143:109906. https://doi.org/10.1016/j.mehy.2020.109906
Fatima N, Saqqur M, Qamar F, Shaukat S, Shuaib A (2020) Impact of COVID-19 on neurological manifestations: an overview of stroke presentation in pandemic. Neurol Sci 41:2675–2679. https://doi.org/10.1007/s10072-020-04637-6
Wang HL, Tang XJ, Fan HY, Luo YH, Song YX, Xu Y, Chen YZ (2020) Potential mechanisms of hemorrhagic stroke in elderly COVID-19 patients. Aging (Albany NY) 12(11):10022–10034. https://doi.org/10.18632/aging.103335
Kalovyrna N, Apokotou O, Boulekou S, Paouri E, Boutou A, Georgopoulos S (2020) A 3’UTR modification of the TNF-alpha mouse gene increases peripheral TNF-alpha and modulates the Alzheimer-like phenotype in 5XFAD mice. Sci Rep 10(1):8670. https://doi.org/10.1038/s41598-020-65378-2
Labandeira-Garcia JL, Garrido-Gil P, Rodriguez-Pallares J, Valenzuela R, Borrajo A, Rodriguez-Perez AI (2014) Brain renin-angiotensin system and dopaminergic cell vulnerability. Front Neuroanat 8:67. https://doi.org/10.3389/fnana.2014.00067
Kehoe PG, Miners S, Love S (2009) Angiotensins in Alzheimer’s disease — friend or foe? Trends Neurosci 32(12):619–628. https://doi.org/10.1016/j.tins.2009.07.006
Bostanciklioglu M (2020) Severe acute respiratory syndrome coronavirus 2 is penetrating to dementia research. Curr Neurovasc Res 17:342–343. https://doi.org/10.2174/1567202617666200522220509
Ennerfelt HE, Lukens JR (2020) The role of innate immunity in Alzheimer’s disease. Immunol Rev 297:225–246. https://doi.org/10.1111/imr.12896
Delgado-Roche L, Mesta F (2020) Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch Med Res 51:384–387. https://doi.org/10.1016/j.arcmed.2020.04.019
Panfoli I (2020) Potential role of endothelial cell surface ectopic redox complexes in COVID-19 disease pathogenesis. Clin Med (Lond) 20(5):e146–e147. https://doi.org/10.7861/clinmed.2020-0252
Nasi A, McArdle S, Gaudernack G, Westman G, Melief C, Rockberg J, Arens R, Kouretas D, Sjölin J, Mangsbo S (2020) Reactive oxygen species as an initiator of toxic innate immune responses in retort to SARS-CoV-2 in an ageing population, consider N-acetylcysteine as early therapeutic intervention. Toxicol Rep 7:768–771. https://doi.org/10.1016/j.toxrep.2020.06.003
Hardas S, Sultana R, Clark A, Beckett T, Szweda L, Murphy M, Butterfield D (2013) Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol 1:80–85. https://doi.org/10.1016/j.redox.2013.01.002
Lima M, Siokas V, Aloizou A, Liampas I, Mentis A, Tsouris Z, Papadimitriou A, Mitsias P, Tsatsakis A, Bogdanos D, Baloyannis S, Dardiotis E (2020) Unraveling the possible routes of SARS-COV-2 Invasion into the central nervous system. Curr Treat Options Neurol 22(11):37. https://doi.org/10.1007/s11940-020-00647-z
Zubair AS, McAlpine LS, Gardin T, Farhadian S, Kuruvilla DE, Spudich S (2020) Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: A Review. JAMA Neurol 77(8):1018–1027. https://doi.org/10.1001/jamaneurol.2020.2065
Politi LS, Salsano E, Grimaldi M (2020) Magnetic resonance imaging alteration of the brain in a patient with coronavirus disease 2019 (COVID-19) and anosmia. JAMA Neurol 77(8):1028–1029. https://doi.org/10.1001/jamaneurol.2020.2125
Chen X, Laurent S, Onur O, Kleineberg N, Fink G, Schweitzer F, Warnke C (2021) A systematic review of neurological symptoms and complications of COVID-19. J Neurol 268:392–402. https://doi.org/10.1007/s00415-020-10067-3
Lim K, Yang S, Kim S, Joo J (2020) Elevation of ACE2 as a SARS-CoV-2 entry receptor gene expression in Alzheimer’s disease. J Infect 81(3):e33–e34. https://doi.org/10.1016/j.jinf.2020.06.072
Reddy S, Hashmi A (2020) Managing diabetes and dementia. Clin Geriatr Med 36(3):419–429. https://doi.org/10.1016/j.cger.2020.04.003
Dubey SK, Lakshmi KK, Krishna KV, Agrawal M, Singhvi G, Saha RN, Saraf S, Saraf S, Shukla R, Alexander A (2020) Insulin mediated novel therapies for the treatment of Alzheimer’s disease. Life Sci 249:117540. https://doi.org/10.1016/j.lfs.2020.117540
Shinohara M, Sato N (2017) Bidirectional interactions between diabetes and Alzheimer’s disease. Neurochem Int 108:296–302. https://doi.org/10.1016/j.neuint.2017.04.020
Ohyagi Y, Miyoshi K, Nakamura N (2019) Therapeutic strategies for Alzheimer’s disease in the view of diabetes mellitus. Adv Exp Med Biol 1128:227–248. https://doi.org/10.1007/978-981-13-3540-2_11
Obukhov A, Stevens B, Prasad R, Li Calzi S, Boulton M, Raizada M, Oudit G, Grant M (2020) SARS-CoV-2 Infections and ACE2: clinical outcomes linked with increased morbidity and mortality in individuals with diabetes. Diabetes 69(9):1875–1886. https://doi.org/10.2337/dbi20-0019
Hollstein T, Schulte D, Schulz J, Glück A, Ziegler A, Bonifacio E, Wendorff M, Franke A, Schreiber S, Bornstein S, Laudes M (2020) Autoantibody-negative insulin-dependent diabetes mellitus after SARS-CoV-2 infection: a case report. Nat Metab 2(10):1021–1024. https://doi.org/10.1038/s42255-020-00281-8
Bardach SH, Gibson AK, Rhodus EK, Jicha GA (2020) Will “social distancing” lead to future “research distancing”: a reflection on COVID-19 impacts on Alzheimer’s disease research. Alzheimers Dement (N Y) 6(1):e12045. https://doi.org/10.1002/trc2.12045
The Editors of Alzheimer’s & Dementia (2020) Alzheimer's disease research enterprise in the era of COVID-19/SARS-CoV-2. Alzheimers Dement 16(4): 587–588. https://doi.org/10.1002/alz.12093
Udeh-Momoh CT, de Jager-Loots CA, Price G, Middleton LT (2020) Transition from physical to virtual visit format for a longitudinal brain aging study, in response to the Covid-19 pandemic. Operationalizing adaptive methods and challenges. Alzheimers Dement (N Y) 6(1):e12010.1002/trc2.12055
Matias-Guiu J, Pytel V, Matías-Guiu J (2020) Death rate due to COVID-19 in Alzheimer’s disease and frontotemporal dementia. J Alzheimers Dis 78(2):537–541. https://doi.org/10.3233/JAD-200940
Shigihara Y, Hoshi H, Poza J, Rodríguez-González V, Gómez C, Kanzawa T (2020) Predicting the outcome of non-pharmacological treatment for patients with dementia-related mild cognitive impairment. Aging (Albany NY). 12(23):24101–24116. https://doi.org/10.18632/aging.202270
Varvel N, Bhaskar K, Kounnas M, Wagner S, Yang Y, Lamb B, Herrup K (2009) NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J Clin Investig 119(12):3692–3702. https://doi.org/10.1172/JCI39716
Aggarwal BB, Sundaram C, Prasad S, Kannappan R (2010) Tocotrienols, the vitamin E of the 21st century: its potential against cancer and other chronic diseases. Biochem Pharmacol 80(11):1613–1631. https://doi.org/10.1016/j.bcp.2010.07.043
Chin KY, Tay SS (2018) A Review on the relationship between tocotrienol and Alzheimer disease. Nutrients 10(7):881. https://doi.org/10.3390/nu10070881
Marinelli R, Torquato P, Bartolini D, Mas-Bargues C, Bellezza G, Gioiello A, Borras C, De Luca A, Fallarino F, Sebastiani B, Mani S, Sidoni A, Viña J, Leri M, Bucciantini M, Nardiello P, Casamenti F, Galli F (2020) Garcinoic acid prevents β-amyloid (Aβ) deposition in the mouse brain. J Biol Chem 295(33):013303. https://doi.org/10.1074/jbc.RA120.013303
Pistollato F, Sumalla Cano S, Elio I, Masias Vergara M, Giampieri F, Battino M (2016) Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr Rev 74(10):624–634. https://doi.org/10.1093/nutrit/nuw023
Daulatzai M (2014) Chronic functional bowel syndrome enhances gut-brain axis dysfunction, neuroinflammation, cognitive impairment, and vulnerability to dementia. Neurochem Res 39(4):624–644. https://doi.org/10.1007/s11064-014-1266-6
Rodriguez-Casado A, Toledano-Díaz A, Toledano A (2017) Defective insulin signalling, mediated by inflammation, connects obesity to Alzheimer disease; relevant pharmacological therapies and Preventive dietary interventions. J Current Alzheimer research 14(8):894–911. https://doi.org/10.2174/1567205014666170316161848
Tosto G, Bird TD, Bennett DA, Boeve BF, Brickman AM, Cruchaga C, Faber K, Foroud TM, Farlow M, Goate AM, Graff-Radford NR, Lantigua R, Manly J, Ottman R, Rosenberg R, Schaid DJ, Schupf N, Stern Y, Sweet RA, Mayeux R (2016) National Institute on Aging Late-Onset Alzheimer Disease/National Cell Repository for Alzheimer Disease (NIA-LOAD/NCRAD) family study group. The role of cardiovascular risk factors and stroke in familial Alzheimer disease. JAMA Neurol 73(10):1231–1237. https://doi.org/10.1001/jamaneurol.2016.2539
Veronese N, Solmi M, Basso C, Smith L, Soysal P (2019) Role of physical activity in ameliorating neuropsychiatric symptoms in Alzheimer disease: a narrative review. Int J Geriatr Psychiatry 34(9):1316–1325. https://doi.org/10.1002/gps.4962
McGough E, Kirk-Sanchez N, Liu-Ambrose T (2017) Integrating health promotion into physical therapy practice to improve brain health and prevent Alzheimer disease. J Neurol Phys Ther 07:S55–S62. https://doi.org/10.1097/NPT.0000000000000181
Fiore V, De Rosa A, Falasca P, Marci M, Guastamacchia E, Licchelli B, Giagulli V, De Pergola G, Poggi A, Triggiani V (2019) Focus on the correlations between Alzheimer’s disease and type 2 diabetes. Endocr Metab Immune Disord Drug Targets 19(5):571–579. https://doi.org/10.2174/1871530319666190311141855
Exalto L, Whitmer R, Kappele L, Biessels G (2012) An update on type 2 diabetes, vascular dementia and Alzheimer’s disease. Exp Gerontol 47(11):858–864. https://doi.org/10.1016/j.exger.2012.07.014
Sardu C, D’Onofrio N, Balestrieri M, Barbieri M, Rizzo M, Messina V, Maggi P, Coppola N, Paolisso G, Marfella R (2020) Outcomes in patients with hyperglycemia affected by COVID-19: can we do more on glycemic control? Diabetes Care 43(7):1408–1415. https://doi.org/10.2337/dc20-0723
García-Arriaza J, Marín M, Merchán-Rubira J, Mascaraque S, Medina M, Ávila J, Hernández F, Esteban M (2020) Tauopathy analysis in P301S mouse model of Alzheimer disease immunized with DNA and MVA poxvirus-based vaccines expressing human full-length 4R2N or 3RC Tau proteins. Vaccines (Basel) 8(1):127. https://doi.org/10.3390/vaccines8010127
Sterner R, Takahashi P, Yu Ballard A (2016) Active vaccines for Alzheimer disease treatment. J Am Med Dir Assoc 17(9):862.e811-865. https://doi.org/10.1016/j.jamda.2016.06.009
Rees J (1981) Clinical interpretation of pharmacokinetic data on dothiepin hydrochloride (Dosulepin, Prothiaden). J Int Med Res 9(2):98–102. https://doi.org/10.1177/030006058100900202
Song G, Yang H, Shen N, Pham P, Brown B, Lin X, Hong Y, Sinu P, Cai J, Li X, Leon M, Gordon MN, Morgan D, Zhang S, Cao C (2020) An immunomodulatory therapeutic vaccine targeting oligomeric amyloid-β. J Alzheimers Dis 77(4):1639–1653. https://doi.org/10.3233/JAD-200413
Acknowledgements
We would like to thank Neurological Sciences for providing this excellent platform for researchers to communicate and learn.
Funding
This study was supported by “Six Talents” Peak Cultivation Funding Project (No.WSW-246); Jiangsu Province, “Thirteenth Five-Year Plan” Special Fund for Science, Education, and Health (No. RCC201807); Provincial academic leader Class B (No. DTRB18008).
Author information
Authors and Affiliations
Contributions
Haili Wang, Juan Lu and Xia Zhao: writing of the manuscript and making of figures. Yingzhu Chen and Jun Zhang: revising the manuscript and accepting responsibility for conduct of research and final approval. All authors contributed to writing and revising of the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors report no conflicts of interest in this work.
Ethical approval
None.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The above article was published online with missing statement. An article note: "Haili Wang, Juan Lu and Xia Zhao contributed equally to this work" has been added.
Haili Wang, Juan Lu and Xia Zhao contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wang, H., Lu, J., Zhao, X. et al. Alzheimer’s disease in elderly COVID-19 patients: potential mechanisms and preventive measures. Neurol Sci 42, 4913–4920 (2021). https://doi.org/10.1007/s10072-021-05616-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-021-05616-1