
New In Silico Insights into the Application of (Hydroxy)Chloroquine with Macrolide Antibiotic Co-Crystals against the SARS-CoV-2 Virus


 and
and
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Assis, L.C.; de Castro, A.A.; de Jesus, J.P.A.; Nepovimova, E.; Kuca, K.; Ramalho, T.C.; La Porta, F.A. Computational evidence for nitro derivatives of quinoline and quinoline N-oxide as low-cost alternative for the treatment of SARS-CoV-2 infection. Sci. Rep. 2021, 11, 6397. [Google Scholar] [CrossRef] [PubMed]
- Arabi, Y.M.; Murthy, S.; Webb, S. COVID-19: A novel coronavirus and a novel challenge for critical care. Intensive Care Med. 2020, 46, 833–836. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, eabb3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, M.D.L.; de Oliveira, K.M.T. Comparative Computational Study of SARS-CoV-2 Receptors Antagonists from Already Approved Drugs 2020. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Rosenberg, E.S.; Dufort, E.M.; Udo, T.; Wilberschied, L.A.; Kumar, J.; Tesoriero, J.; Weinberg, P.; Kirkwood, J.; Muse, A.; DeHovitz, J.; et al. Association of Treatment with Hydroxychloroquine or Azithromycin with In-Hospital Mortality in Patients with COVID-19 in New York State. JAMA 2020, 323, 2493–2502. [Google Scholar] [CrossRef] [PubMed]
- Fintelman-Rodrigues, N.; Sacramento, C.Q.; Lima, C.R.; da Silva, F.S.; Ferreira, A.C.; Mattos, M.; de Freitas, C.S.; Soares, V.C.; da Gomes Dias, S.S.; Temerozo, J.R.; et al. Atazanavir inhibits SARS-CoV-2 replication and pro-inflammatory cytokine production. bioRxiv 2020, 64, e00825-20. [Google Scholar]
- Geleris, J.; Sun, Y.; Platt, J.; Zucker, J.; Baldwin, M.; Hripcsak, G.; Labella, A.; Manson, D.K.; Kubin, C.; Barr, R.G.; et al. Observational Study of Hydroxychloroquine in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2020, 382, 2411–2418. [Google Scholar] [CrossRef]
- Hung, I.F.-N.; Lung, K.-C.; Tso, E.Y.-K.; Liu, R.; Chung, T.W.-H.; Chu, M.-Y.; Ng, Y.-Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 2020, 395, 1695–1704. [Google Scholar] [CrossRef]
- Nikam, V.J.; Patil, S.B. Pharmaceutical cocrystals of nebivolol hydrochloride with enhanced solubility. J. Cryst. Growth 2020, 534, 125488. [Google Scholar] [CrossRef]
- Steed, J.W. The role of co-crystals in pharmaceutical design. Trends Pharmacol. Sci. 2013, 34, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Maheshwari, C.; Jayasankar, A.; Khan, N.A.; Amidon, G.E.; Rodríguez-Hornedo, N. Factors that influence the spontaneous formation of pharmaceutical cocrystals by simply mixing solid reactants. CrystEngComm 2009, 11, 493–500. [Google Scholar] [CrossRef]
- Leyssens, T.; Tumanova, N.; Robeyns, K.; Candoni, N.; Veesler, S. Solution cocrystallization, an effective tool to explore the variety of cocrystal systems: Caffeine/dicarboxylic acid cocrystals. CrystEngComm 2014, 16, 9603–9611. [Google Scholar] [CrossRef]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef] [PubMed]
- Duggirala, N.K.; Perry, M.L.; Almarsson, Ö.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medicines. Chem. Commun. 2016, 52, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Hennekens, C.H.; Rane, M.; Solano, J.; Alter, S.; Johnson, H.; Krishnaswamy, S.; Shih, R.; Maki, D.; DeMets, D.L. Updates on Hydroxychloroquine in Prevention and Treatment of COVID-19. Am. J. Med. 2022, 135, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Kirst, H.A. Introduction to the Macrolide Antibiotics BT—Macrolide Antibiotics; Schönfeld, W., Kirst, H.A., Eds.; Birkhäuser Basel: Basel, Switzerland, 2002; pp. 1–13. ISBN 978-3-0348-8105-0. [Google Scholar]
- Mcguire, J.M.; Bunch, R.L.; Anderson, R.C.; Boaz, H.E.; Flynn, E.H.; Powell, H.M.; Smith, J.W. Ilotycin, a new antibiotic. Antibiot. Chemother. 1952, 2, 281–283. [Google Scholar]
- Chico, R.M.; Pittrof, R.; Greenwood, B.; Chandramohan, D. Azithromycin-chloroquine and the intermittent preventive treatment of malaria in pregnancy. Malar. J. 2008, 7, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B 2002, 58, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Arsic, B.; Awan, A.; Brennan, R.J.; Aguilar, J.A.; Ledder, R.; McBain, A.J.; Regan, A.C.; Barber, J. Theoretical and experimental investigation on clarithromycin, erythromycin A and azithromycin and descladinosyl derivatives of clarithromycin and azithromycin with 3-O substitution as anti-bacterial agents. Med. Chem. Commun. 2014, 5, 1347–1354. [Google Scholar] [CrossRef]
- Kumar, V.; Tan, K.-P.; Wang, Y.-M.; Lin, S.-W.; Liang, P.-H. Identification, synthesis and evaluation of SARS-CoV and MERS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2016, 24, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Verschueren, K.H.G.; Anand, K.; Shen, J.; Yang, M.; Xu, Y.; Rao, Z.; Bigalke, J.; Heisen, B.; Mesters, J.R.; et al. pH-dependent Conformational Flexibility of the SARS-CoV Main Proteinase (Mpro) Dimer: Molecular Dynamics Simulations and Multiple X-ray Structure Analyses. J. Mol. Biol. 2005, 354, 25–40. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. (3CL pro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Báez-Santos, Y.M.; St. John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Yazdany, J.; Kim, A.H.J. Use of Hydroxychloroquine and Chloroquine during the COVID-19 Pandemic: What Every Clinician Should Know. Ann. Intern. Med. 2020, 172, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Frisch, J.M.; Trucks, W.G.; Schlegel, B.H.; Scuseria, E.G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.H.; et al. Gaussian 09 (Revision A02); Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.C.; dos Pires, M.S.; de Castro, A.A.; da Cunha, E.F.F.; Caetano, M.S.; Ramalho, T.C. Molecular insight into the inhibition mechanism of plant and rat 4-hydroxyphenylpyruvate dioxygenase by molecular docking and DFT calculations. Med. Chem. Res. 2015, 24, 3958–3971. [Google Scholar] [CrossRef]
- Guimaraes, A.P.; Oliveira, A.A.; da Cunha, E.F.F.; Ramalho, T.C.; Franco, T.C.C. Analysis of Bacillus anthracis nucleoside hydrolase via in silico docking with inhibitors and molecular dynamics simulation. J. Mol. Model. 2011, 17, 2939–2951. [Google Scholar] [CrossRef] [PubMed]
- Matos, K.S.; Mancini, D.T.; da Cunha, E.F.F.; Kuca, K.; Franca, T.C.C.; Ramalho, T.C. Molecular Aspects of the Reactivation Process of Acetylcholinesterase Inhibited by Cyclosarin. J. Braz. Chem. Soc. 2011, 22, 1999–2004. [Google Scholar] [CrossRef] [Green Version]
- da Cunha, E.F.F.; Barbosa, E.F.; Oliveira, A.A.; Ramalho, T.C. Molecular Modeling of Mycobacterium Tuberculosis DNA Gyrase and its Molecular Docking Study with Gatifloxacin Inhibitors. J. Biomol. Struct. Dyn. 2010, 27, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Souza, T.C.S.; Josa, D.; Ramalho, T.C.; Caetano, M.S.; da Cunha, E.F.F. Molecular modelling of Mycobacterium tuberculosis acetolactate synthase catalytic subunit and its molecular docking study with inhibitors. Mol. Simul. 2008, 34, 707–713. [Google Scholar] [CrossRef]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; van Gunsteren, W.F. The GROMOS Biomolecular Simulation Program Package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Deepa, G.; Sivakumar, K.C.; Sajeevan, T.P. Molecular simulation and in vitro evaluation of chitosan nanoparticles as drug delivery systems for the controlled release of anticancer drug cytarabine against solid tumours. 3 Biotech 2018, 8, 493. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Fernandes, R.F.; Stroppa, P.H.F.; Ferreira, G.R.; da Silva, A.D.; Edwards, H.G.M.; de Oliveira, L.F.C. Vibrational spectroscopic study of some quinoline derivatives. Vib. Spectrosc. 2016, 86, 128–133. [Google Scholar] [CrossRef]
- Adeli, E. Preparation and evaluation of azithromycin binary solid dispersions using various polyethylene glycols for the improvement of the drug solubility and dissolution rate. Braz. J. Pharm. Sci. 2016, 52, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Dey, S. Plasmon Enhanced Hybrid Photovoltaics. In Imerging Research in Science and Engineering Based on Advanced Experimental and Computational Strategies. Engineering Materials; La Porta, F.A., Taft, C.A., Eds.; Springer: Cham, Switzerland, 2020; ISBN 978-3-030-31402-6. [Google Scholar]
- La Porta, F.A.; Ramalho, T.C.; Santiago, R.T.; Rocha, M.V.J.; da Cunha, E.F.F. Orbital Signatures as a Descriptor of Regioselectivity and Chemical Reactivity: The Role of the Frontier Orbitals on 1,3-Dipolar Cycloadditions. J. Phys. Chem. A 2011, 115, 824–833. [Google Scholar] [CrossRef] [PubMed]
- da Silva, R.R.; Ramalho, T.C.; Santos, J.M.; Figueroa-Villar, J.D. On the Limits of Highest-Occupied Molecular Orbital Driven Reactions: The Frontier Effective-for-Reaction Molecular Orbital Concept. J. Phys. Chem. A 2006, 110, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Yonezawa, T.; Shingu, H. A Molecular Orbital Theory of Reactivity in Aromatic Hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Weinhold, F. Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure. J. Mol. Struct. Theochem 1988, 169, 41–62. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Yang, S.; Chen, S.J.; Hsu, M.F.; Wu, J.D.; Tseng, C.T.K.; Liu, Y.F.; Chen, H.C.; Kuo, C.W.; Wu, C.S.; Chang, L.W.; et al. Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem. 2006, 49, 4971–4980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Liu, Y.; Zhang, W.; Wei, P.; Huang, C.; Pei, J.; Yuan, Y.; Lai, L. Isatin compounds as noncovalent SARS coronavirus 3C-like protease inhibitors. J. Med. Chem. 2006, 49, 3440–3443. [Google Scholar] [CrossRef] [PubMed]
- Cardoso Gajo, G.; Rodrigues Silva, D.; Barigye, S.J.; da Cunha, E.F.F. Multi-objective Optimization of Benzamide Derivatives as Rho Kinase Inhibitors. Mol. Inform. 2018, 37, 1700080. [Google Scholar] [CrossRef]
- La Porta, F.A.; Taft, C. Emerging Research in Science and Engineering Based on Advanced Experimental and Computational Strategies; Springer Nature: Berlin/Heidelberg, Germany, 2020; ISBN 978-3-030-31402-6. [Google Scholar]
- La Porta, F.A.; Taft, C.A. (Eds.) Functional Properties of Advanced Engineering Materials and Biomolecules, 1st ed.; Springer International Publishing: Cham, Switzerland, 2021; ISBN 978-3-030-62225-1. [Google Scholar]
- Ramalho, T.C.; de Castro, A.A.; Silva, D.R.; Silva, M.C.; Franca, T.C.C.; Bennion, B.J.; Kuca, K. Computational Enzymology and Organophosphorus Degrading Enzymes: Promising Approaches toward Remediation Technologies of Warfare Agents and Pesticides. Curr. Med. Chem. 2016, 23, 1041–1061. [Google Scholar] [CrossRef] [PubMed]
- de Castro, A.A.; Prandi, I.G.; Kuca, K.; Ramalho, T.C. Organophosphorus degrading enzymes: Molecular basis and perspectives for enzymatic bioremediation of agrochemicals. Cienc. Agrotecnologia 2017, 41, 471–482. [Google Scholar]
- Durrant, J.D.; Kochanek, S.E.; Casalino, L.; Ieong, P.U.; Dommer, A.C.; Amaro, R.E. Mesoscale All-Atom Influenza Virus Simulations Suggest New Substrate Binding Mechanism. ACS Cent. Sci. 2020, 6, 189–196. [Google Scholar] [CrossRef] [PubMed]








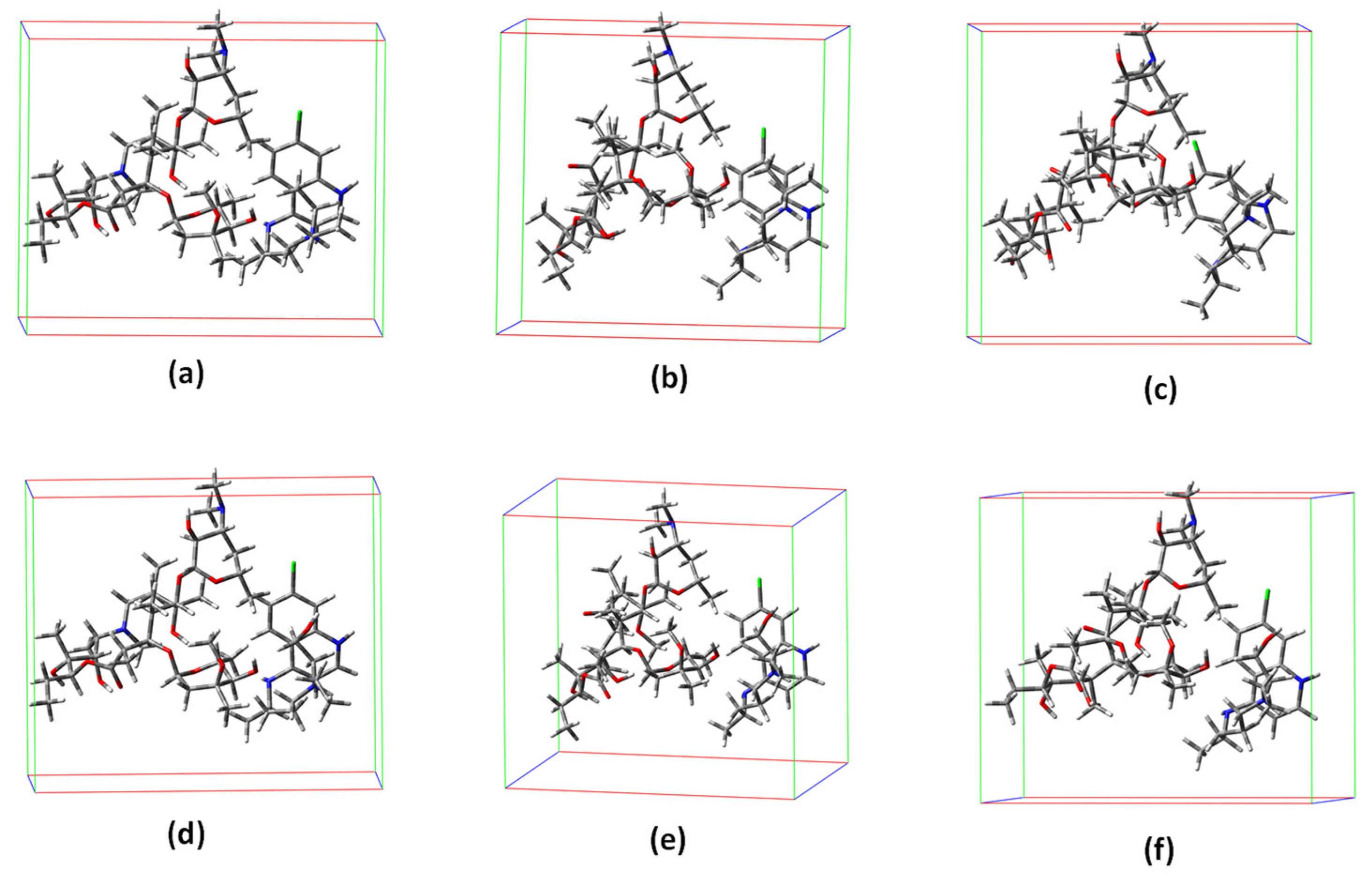{kind=link}
{kind=link}
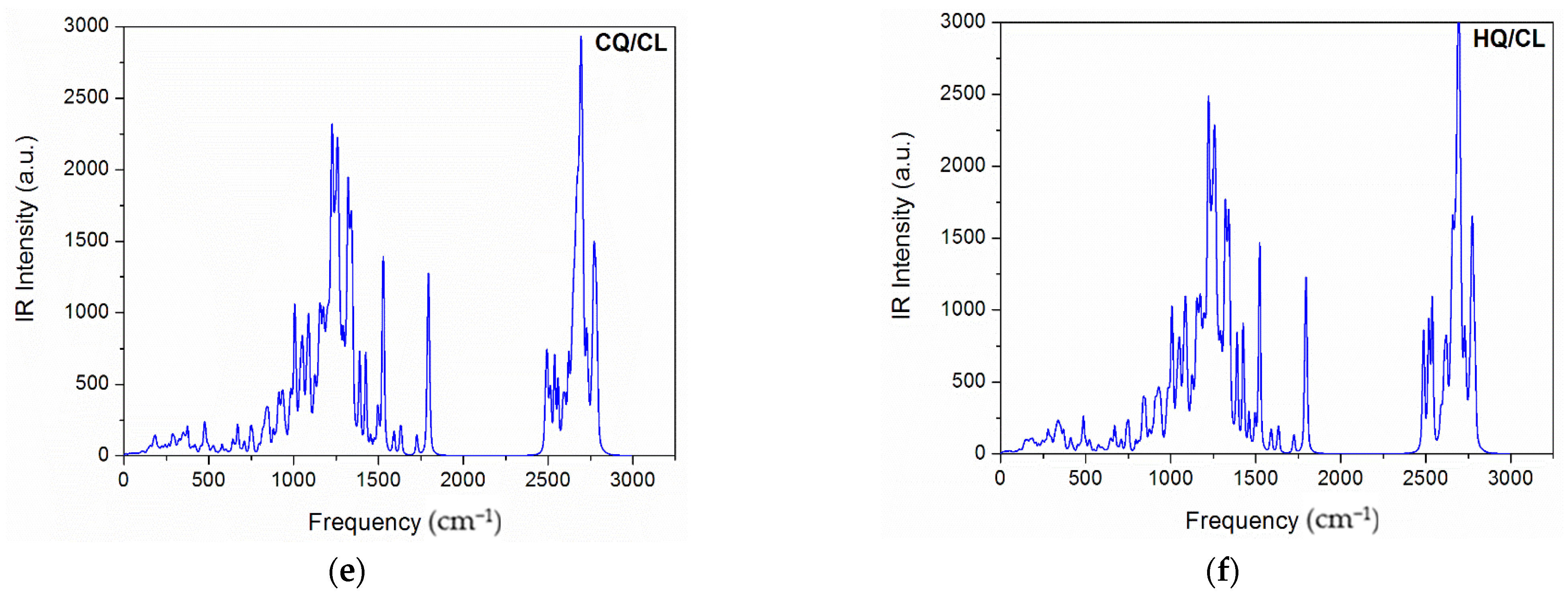{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co-Crystal | ΔE (kcal mol−1) | H° (kcal mol−1) | S° cal (mol K)−1 | CV cal (mol K)−1 |
|---|---|---|---|---|
| CQ/AZ | 97.2 | 932.4 | 459.3 | 333.9 |
| CQ/ER | 48.9 | 929.2 | 462.3 | 336.1 |
| CQ/CL | 52.0 | 946.8 | 476.3 | 342.1 |
| HQ/AZ | 52.3 | 935.2 | 469.3 | 337.5 |
| HQ/ER | 0.0 | 931.6 | 471.5 | 340.2 |
| HQ/CL | 7.6 | 949.5 | 482.5 | 345.6 |
| Co-Crystal | Eg (eV) | EB (eV) | ECT | µ (D) | η (eV) | S (eV−1) | x (eV) | ω |
|---|---|---|---|---|---|---|---|---|
| CQ/AZ | 1.22 | 0.26 | 0.009 | −1.03 | 0.61 | 0.82 | 1.03 | 0.43 |
| CQ/ER | 1.26 | 0.07 | 0.141 | −1.02 | 0.63 | 0.79 | 1.02 | 0.44 |
| CQ/CL | 1.46 | 0.47 | 0.337 | −0.94 | 0.73 | 0.68 | 0.94 | 0.52 |
| HQ/AZ | 1.44 | 0.26 | 0.019 | −1.10 | 0.72 | 0.69 | 1.10 | 0.46 |
| HQ/ER | 1.44 | 0.26 | 0.632 | −1.13 | 0.72 | 0.70 | 1.14 | 0.45 |
| HQ/CL | 1.45 | 0.28 | 0.350 | −1.12 | 0.73 | 0.69 | 1.12 | 0.46 |
| Co-Crystal | Interaction Energy (kcal mol−1) | Toxicity (LD50) |
|---|---|---|
| AZ | −213.2 | 2.54 mol kg−1 |
| CL | −205.0 | 2.72 mol kg−1 |
| CQ/AZ | −275.3 | 2.74 mol kg−1 |
| CQ/CL | −240.9 | 2.75 mol kg−1 |
| CQ | −141.7 | 2.95 mol kg−1 |
| CQ/ER | −229.8 | 2.75 mol kg−1 |
| ER | −213.1 | 2.23 mol kg−1 |
| HQ/AZ | −280.7 | 2.74 mol kg−1 |
| HQ/CL | −259.6 | 2.75 mol kg−1 |
| HQ | −162.6 | 2.63 mol kg−1 |
| HQ/ER | −275.7 | 2.75 mol kg−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Castro, A.A.; Assis, L.C.; da Cunha, E.F.F.; Ramalho, T.C.; La Porta, F.A. New In Silico Insights into the Application of (Hydroxy)Chloroquine with Macrolide Antibiotic Co-Crystals against the SARS-CoV-2 Virus. COVID 2022, 2, 230-243. https://doi.org/10.3390/covid2030018
de Castro AA, Assis LC, da Cunha EFF, Ramalho TC, La Porta FA. New In Silico Insights into the Application of (Hydroxy)Chloroquine with Macrolide Antibiotic Co-Crystals against the SARS-CoV-2 Virus. COVID. 2022; 2(3):230-243. https://doi.org/10.3390/covid2030018
Chicago/Turabian Stylede Castro, Alexandre A., Letícia C. Assis, Elaine F. F. da Cunha, Teodorico C. Ramalho, and Felipe A. La Porta. 2022. "New In Silico Insights into the Application of (Hydroxy)Chloroquine with Macrolide Antibiotic Co-Crystals against the SARS-CoV-2 Virus" COVID 2, no. 3: 230-243. https://doi.org/10.3390/covid2030018