
Immune Signature of COVID-19: In-Depth Reasons and Consequences of the Cytokine Storm

 , , , , ,
, , , , ,  and
and 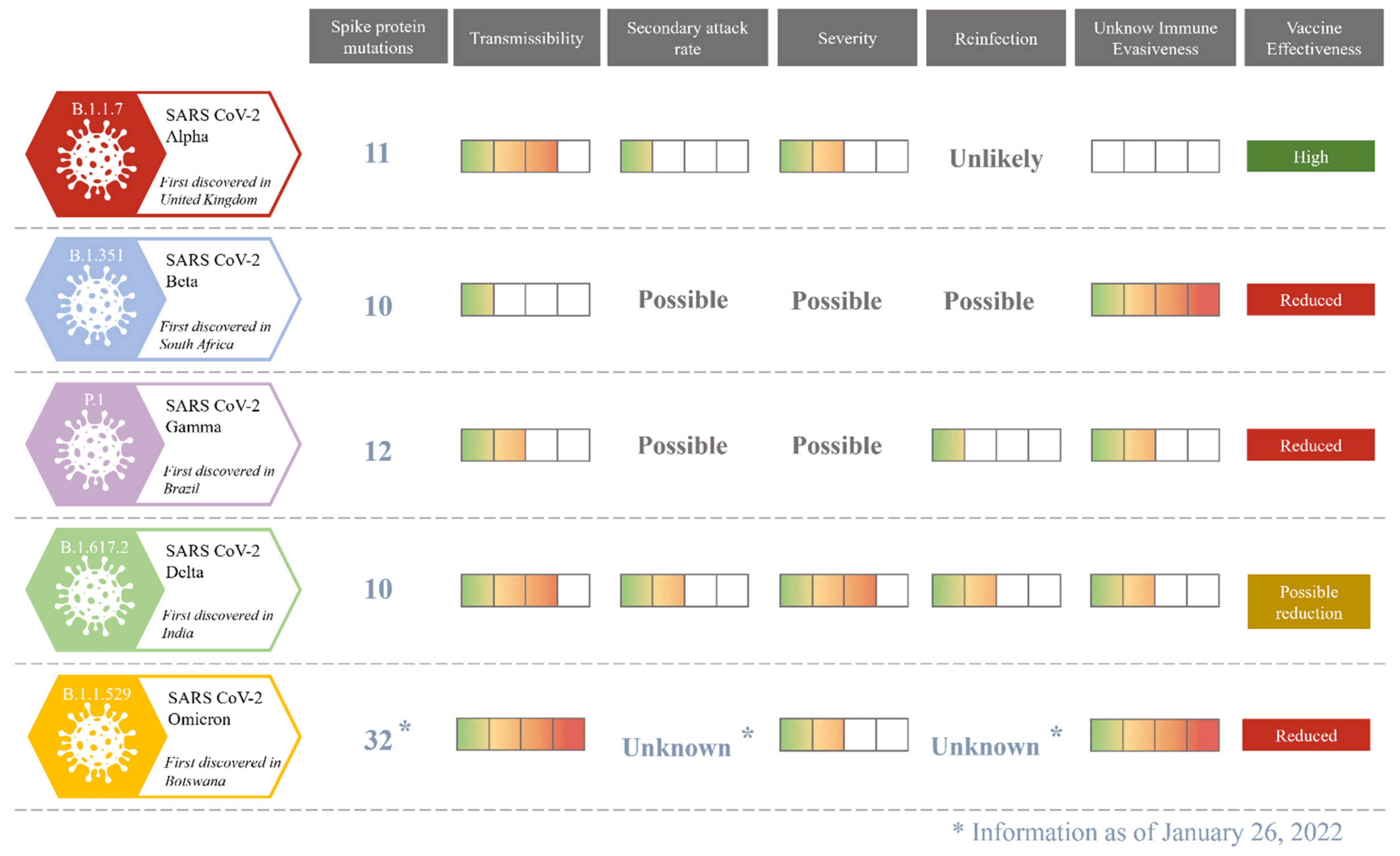{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Virus Molecular Variability
3. COVID-19 Transmission
4. COVID-19 Clinical Manifestations
4.1. Respiratory Manifestations
4.2. Extra-Respiratory Manifestations
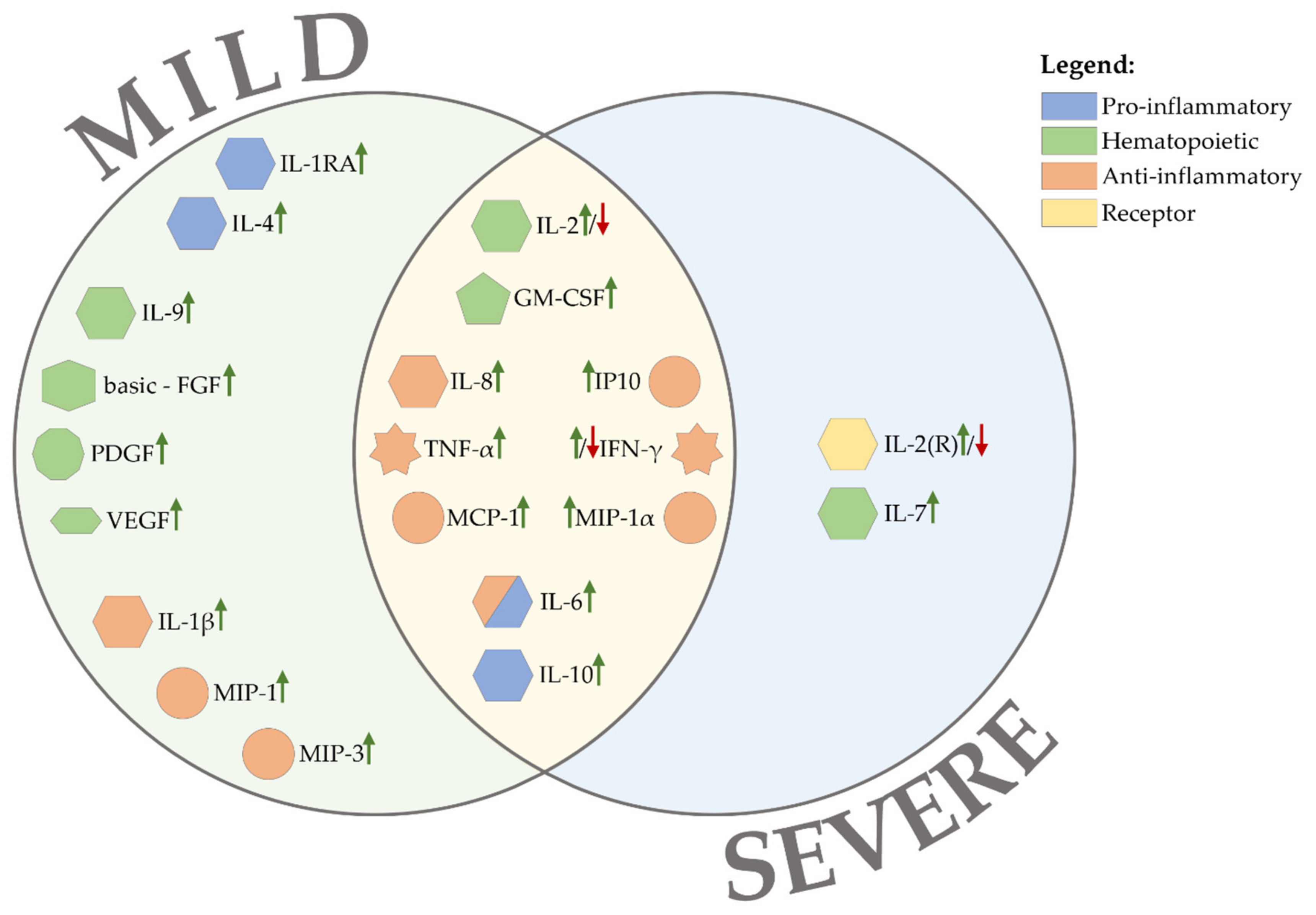
5. Cytokine Profile during SARS-CoV-2
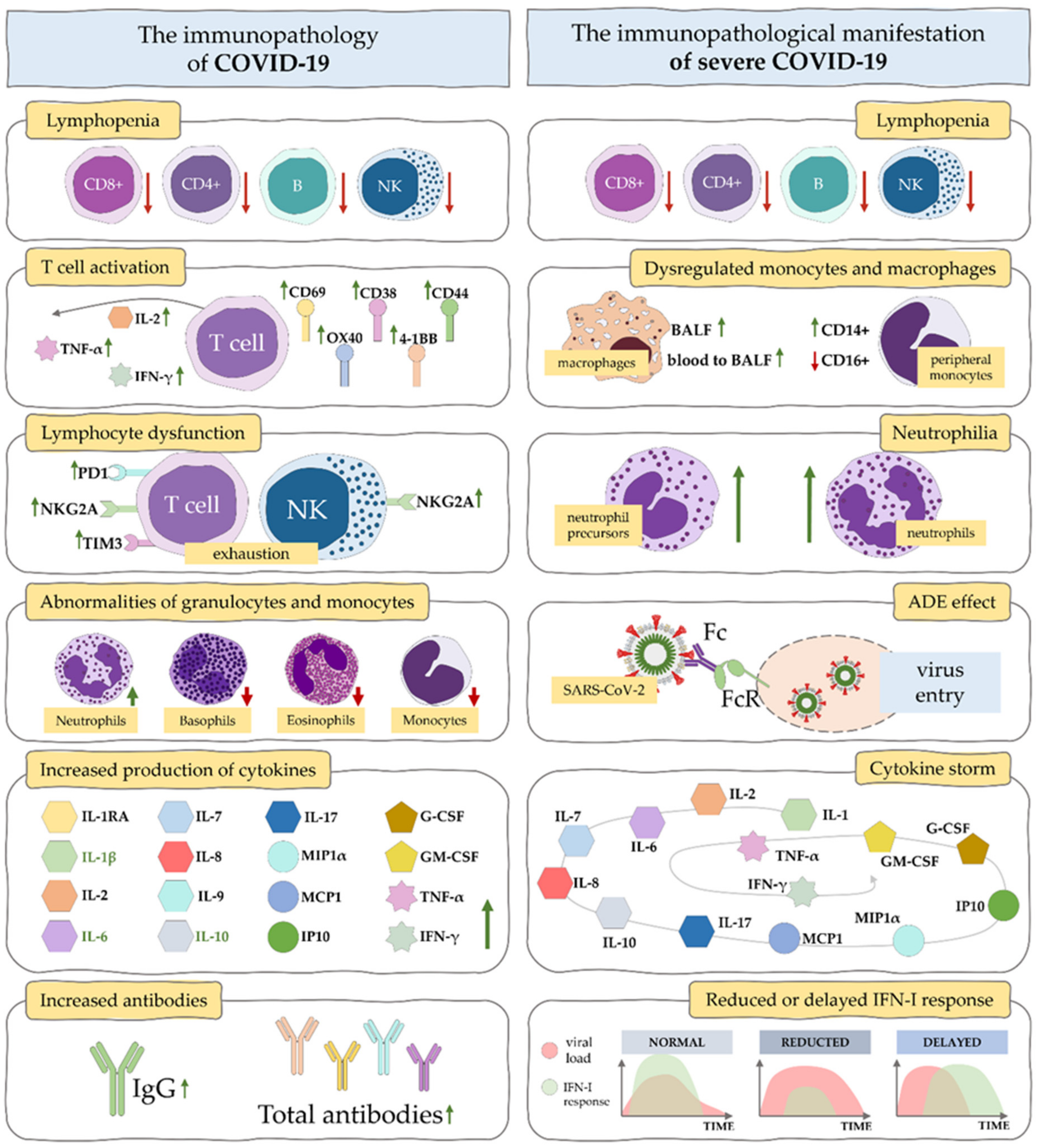
5.1. Signalling Pathways for Cytokine Storm in COVID-19
5.1.1. SARS-CoV-2 Viral Entry
5.1.2. Molecular Signaling Pathways Activated during Cytokine Storm in COVID-19
5.2. Possible Consequences of Cytokine Storm in COVID-19
5.3. Cytokine Storm as a Potential Biomarker in COVID-19 Progression
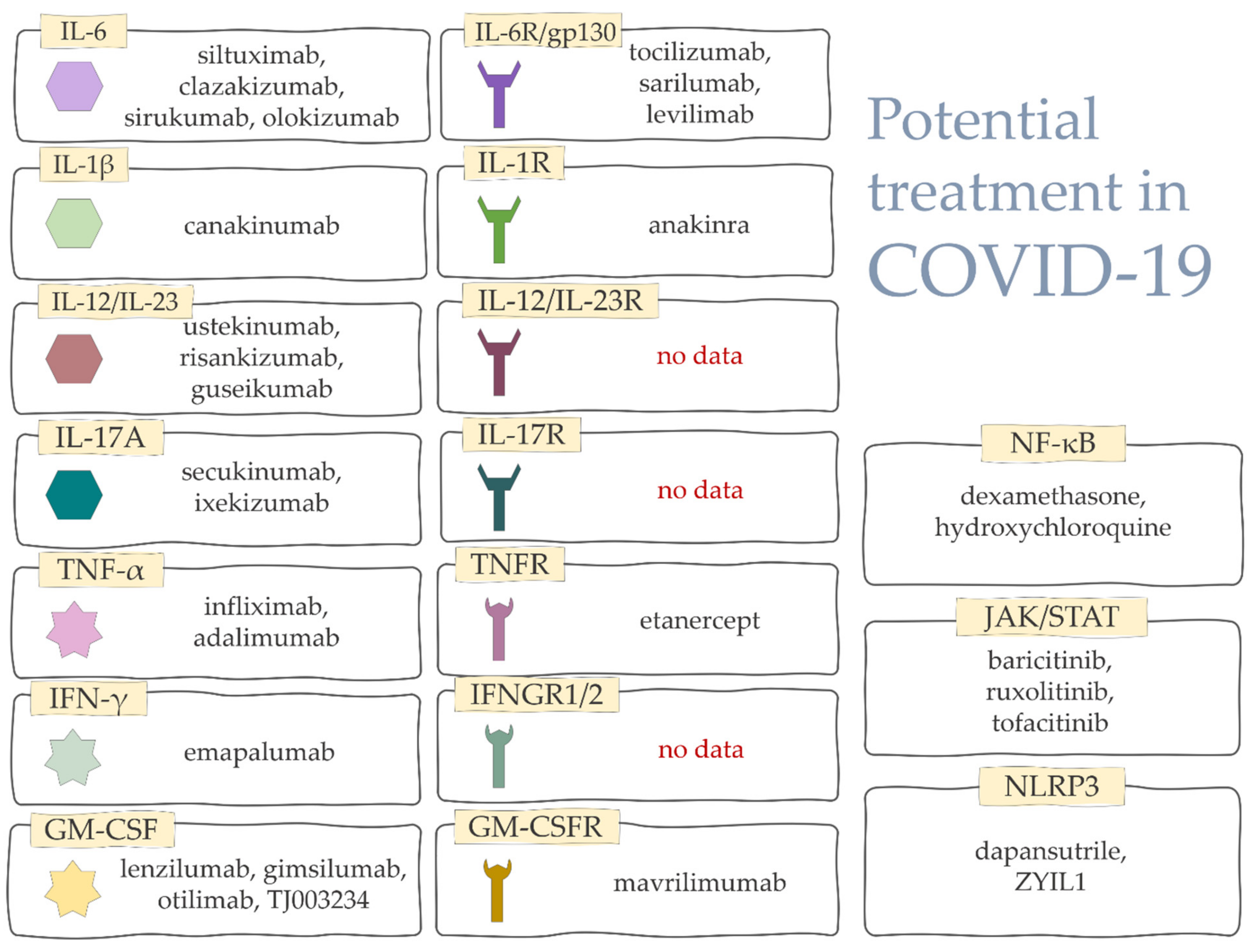
6. Biologic Drugs Used in the Treatment of COVID-CS
6.1. Recombinant Cytokines
6.2. Inhibition of Selected Cytokines
6.3. Blocking Signaling Pathways
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Niedźwiedzka-Rystwej, P.; Grywalska, E.; Hrynkiewicz, R.; Bębnowska, D.; Wołącewicz, M.; Majchrzak, A.; Parczewski, M. Interplay between Neutrophils, NETs and T-Cells in SARS-CoV-2 Infection—A Missing Piece of the Puzzle in the COVID-19 Pathogenesis? Cells 2021, 10, 1817. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 7 January 2022).
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the Alert for Cytokine Storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020, 72, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Petrosillo, N.; Viceconte, G.; Ergonul, O.; Ippolito, G.; Petersen, E. COVID-19, SARS and MERS: Are They Closely Related? Clin. Microbiol. Infect. 2020, 26, 729–734. [Google Scholar] [CrossRef]
- La Rosée, P.; Horne, A.; Hines, M.; von Bahr Greenwood, T.; Machowicz, R.; Berliner, N.; Birndt, S.; Gil-Herrera, J.; Girschikofsky, M.; Jordan, M.B.; et al. Recommendations for the Management of Hemophagocytic Lymphohistiocytosis in Adults. Blood 2019, 133, 2465–2477. [Google Scholar] [CrossRef] [Green Version]
- Ravelli, A.; Minoia, F.; Davì, S.; Horne, A.; Bovis, F.; Pistorio, A.; Aricò, M.; Avcin, T.; Behrens, E.M.; De Benedetti, F.; et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016, 68, 566–576. [Google Scholar] [CrossRef] [Green Version]
- Zachariah, P.; Johnson, C.L.; Halabi, K.C.; Ahn, D.; Sen, A.I.; Fischer, A.; Banker, S.L.; Giordano, M.; Manice, C.S.; Diamond, R.; et al. Epidemiology, Clinical Features, and Disease Severity in Patients With Coronavirus Disease 2019 (COVID-19) in a Children’s Hospital in New York City, New York. JAMA Pediatr. 2020, 174, e202430. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine Release Syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Mangalmurti, N.; Hunter, C.A. Cytokine Storms: Understanding COVID-19. Immunity 2020, 53, 19–25. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine Elevation in Severe and Critical COVID-19: A Rapid Systematic Review, Meta-Analysis, and Comparison with Other Inflammatory Syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Chen, L.Y.C.; Quach, T.T.T. COVID-19 Cytokine Storm Syndrome: A Threshold Concept. Lancet Microbe 2021, 2, e49–e50. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal Characteristics of Lymphocyte Responses and Cytokine Profiles in the Peripheral Blood of SARS-CoV-2 Infected Patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical Spectrum. Available online: https://www.covid19treatmentguidelines.nih.gov/overview/clinical-spectrum/ (accessed on 10 March 2022).
- Yang, L.; Liu, S.; Liu, J.; Zhang, Z.; Wan, X.; Huang, B.; Chen, Y.; Zhang, Y. COVID-19: Immunopathogenesis and Immunotherapeutics. Signal Transduct. Target. Ther. 2020, 5, 128. [Google Scholar] [CrossRef] [PubMed]
- Gallo Marin, B.; Aghagoli, G.; Lavine, K.; Yang, L.; Siff, E.J.; Chiang, S.S.; Salazar-Mather, T.P.; Dumenco, L.; Savaria, M.C.; Aung, S.N.; et al. Predictors of COVID-19 Severity: A Literature Review. Rev. Med. Virol. 2021, 31, e2146. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants (accessed on 10 February 2022).
- Maslo, C.; Friedland, R.; Toubkin, M.; Laubscher, A.; Akaloo, T.; Kama, B. Characteristics and Outcomes of Hospitalized Patients in South Africa during the COVID-19 Omicron Wave Compared with Previous Waves. JAMA 2022, 327, 583–584. [Google Scholar] [CrossRef]
- Wei, C.; Shan, K.-J.; Wang, W.; Zhang, S.; Huan, Q.; Qian, W. Evidence for a Mouse Origin of the SARS-CoV-2 Omicron Variant. J. Genet. Genom. 2021, 48, 1111–1121. [Google Scholar] [CrossRef]
- Murphy, H.L.; Ly, H. Understanding the Prevalence of SARS-CoV-2 (COVID-19) Exposure in Companion, Captive, Wild, and Farmed Animals. Virulence 2021, 12, 2777–2786. [Google Scholar] [CrossRef]
- Madewell, Z.J.; Yang, Y.; Longini, I.M.; Halloran, M.E.; Dean, N.E. Factors Associated with Household Transmission of SARS-CoV-2: An Updated Systematic Review and Meta-Analysis. JAMA Netw. Open 2021, 4, e2122240. [Google Scholar] [CrossRef]
- Farrag, M.A.; Amer, H.M.; Bhat, R.; Hamed, M.E.; Aziz, I.M.; Mubarak, A.; Dawoud, T.M.; Almalki, S.G.; Alghofaili, F.; Alnemare, A.K.; et al. SARS-CoV-2: An Overview of Virus Genetics, Transmission, and Immunopathogenesis. Int. J. Environ. Res. Public Health 2021, 18, 6312. [Google Scholar] [CrossRef]
- Oran, D.P.; Topol, E.J. The Proportion of SARS-CoV-2 Infections That Are Asymptomatic. Ann. Intern. Med. 2021, 173, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Sah, P.; Fitzpatrick, M.C.; Zimmer, C.F.; Abdollahi, E.; Juden-Kelly, L.; Moghadas, S.M.; Singer, B.H.; Galvani, A.P. Asymptomatic SARS-CoV-2 Infection: A Systematic Review and Meta-Analysis. Proc. Natl. Acad. Sci. USA 2021, 118, e2109229118. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-U.; Kim, M.-J.; Ra, S.H.; Lee, J.; Bae, S.; Jung, J.; Kim, S.-H. Clinical Characteristics of Asymptomatic and Symptomatic Patients with Mild COVID-19. Clin. Microbiol. Infect. 2020, 26, 948.e1–948.e3. [Google Scholar] [CrossRef] [PubMed]
- De Souza, T.H.; Nadal, J.A.; Nogueira, R.J.N.; Pereira, R.M.; Brandão, M.B. Clinical Manifestations of Children with COVID-19: A Systematic Review. Pediatr. Pulmonol. 2020, 55, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Barek, M.A.; Aziz, M.A.; Islam, M.S. Impact of Age, Sex, Comorbidities and Clinical Symptoms on the Severity of COVID-19 Cases: A Meta-Analysis with 55 Studies and 10014 Cases. Heliyon 2020, 6, e05684. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of Angiotensin-Converting Enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-Converting Enzyme 2 Is a Functional Receptor for the SARS Coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Mossel, E.C.; Wang, J.; Jeffers, S.; Edeen, K.E.; Wang, S.; Cosgrove, G.P.; Funk, C.J.; Manzer, R.; Miura, T.A.; Pearson, L.D.; et al. SARS-CoV Replicates in Primary Human Alveolar Type II Cell Cultures but Not in Type I-like Cells. Virology 2008, 372, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.D.; Harris, C.; Cain, J.K.; Hummer, C.; Goyal, H.; Perisetti, A. Pulmonary and Extra-Pulmonary Clinical Manifestations of COVID-19. Front. Med. 2020, 7, 526. [Google Scholar] [CrossRef]
- Zhang, X.; Li, S.; Niu, S. ACE2 and COVID-19 and the Resulting ARDS. Postgrad. Med. J. 2020, 96, 403–407. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, W.; Li, J.; Chen, L.; Wang, Y.; Zhang, L.; Yu, L. Clinical Features and Progression of Acute Respiratory Distress Syndrome in Coronavirus Disease 2019. medRxiv 2020. [Google Scholar] [CrossRef]
- Tzotzos, S.J.; Fischer, B.; Fischer, H.; Zeitlinger, M. Incidence of ARDS and Outcomes in Hospitalized Patients with COVID-19: A Global Literature Survey. Crit. Care 2020, 24, 516. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.G.; Qin, L.; Puah, S.H. COVID-19 Acute Respiratory Distress Syndrome (ARDS): Clinical Features and Differences from Typical Pre-COVID-19 ARDS. Med. J. Aust. 2020, 213, 54–56.e1. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, C.; Suarez-Sipmann, F.; Mellado-Artigas, R.; Hernández, M.; Gea, A.; Arruti, E.; Aldecoa, C.; Martínez-Pallí, G.; Martínez-González, M.A.; Slutsky, A.S.; et al. Clinical Features, Ventilatory Management, and Outcome of ARDS Caused by COVID-19 Are Similar to Other Causes of ARDS. Intensive Care Med. 2020, 46, 2200–2211. [Google Scholar] [CrossRef]
- Reghunathan, R.; Jayapal, M.; Hsu, L.-Y.; Chng, H.-H.; Tai, D.; Leung, B.P.; Melendez, A.J. Expression Profile of Immune Response Genes in Patients with Severe Acute Respiratory Syndrome. BMC Immunol. 2005, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Wang, B.; Mao, J. The Pathogenesis and Treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Wong, C.K.; Lam, C.W.K.; Wu, A.K.L.; Ip, W.K.; Lee, N.L.S.; Chan, I.H.S.; Lit, L.C.W.; Hui, D.S.C.; Chan, M.H.M.; Chung, S.S.C.; et al. Plasma Inflammatory Cytokines and Chemokines in Severe Acute Respiratory Syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.; Ross, R.; Frydas, I.; Kritas, S. Induction of Pro-Inflammatory Cytokines (IL-1 and IL-6) and Lung Inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-Inflammatory Strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 327–331. [Google Scholar] [CrossRef]
- Mehta, P.; Porter, J.; Manson, J.; Isaacs, J.; Openshaw, P.; McInnes, I.; Summers, C.; Chambers, R. Therapeutic Blockade of Granulocyte Macrophage Colony-Stimulating Factor in COVID-19-Associated Hyperinflammation: Challenges and Opportunities. Lancet Respir. Med. 2020, 8, 822–830. [Google Scholar] [CrossRef]
- Boretti, A.; Banik, B.K. Intravenous Vitamin C for Reduction of Cytokines Storm in Acute Respiratory Distress Syndrome. Pharma Nutr. 2020, 12, 100190. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary Manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Lai, C.-C.; Ko, W.-C.; Lee, P.-I.; Jean, S.-S.; Hsueh, P.-R. Extra-Respiratory Manifestations of COVID-19. Int. J. Antimicrob. Agents 2020, 56, 106024. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewska, H.; Skonieczna-Żydecka, K.; Parczewski, M.; Niścigorska-Olsen, J.; Karpińska, E.; Hornung, M.; Jurczyk, K.; Witak-Jędra, M.; Laurans, Ł.; Maciejewska, K.; et al. Hepatotropic Properties of SARS-CoV-2-Preliminary Results of Cross-Sectional Observational Study from the First Wave COVID-19 Pandemic. J. Clin. Med. 2021, 10, 672. [Google Scholar] [CrossRef] [PubMed]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.-D.; Sacco, C.; Bertuzzi, A.; et al. Venous and Arterial Thromboembolic Complications in COVID-19 Patients Admitted to an Academic Hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of Thrombotic Complications in Critically Ill ICU Patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Ahmed, S.; Zimba, O.; Gasparyan, A.Y. Thrombosis in Coronavirus Disease 2019 (COVID-19) through the Prism of Virchow’s Triad. Clin. Rheumatol. 2020, 39, 2529–2543. [Google Scholar] [CrossRef]
- Maier, C.L.; Truong, A.D.; Auld, S.C.; Polly, D.M.; Tanksley, C.-L.; Duncan, A. COVID-19-Associated Hyperviscosity: A Link between Inflammation and Thrombophilia? Lancet 2020, 395, 1758–1759. [Google Scholar] [CrossRef]
- Gando, S.; Wada, T. Thromboplasminflammation in COVID-19 Coagulopathy: Three Viewpoints for Diagnostic and Therapeutic Strategies. Front. Immunol. 2021, 12, 649122. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulos, A.C. The Management of Venous Thromboembolism in Hospitalized Patients with COVID-19. Blood Adv. 2020, 4, 4028. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.L.; Vogler, T.O.; Moore, E.E.; Moore, H.B.; Wohlauer, M.V.; Urban, S.; Nydam, T.L.; Moore, P.K.; McIntyre, R.C. Fibrinolysis Shutdown Correlation with Thromboembolic Events in Severe COVID-19 Infection. J. Am. Coll. Surg. 2020, 231, 193–203.e1. [Google Scholar] [CrossRef] [PubMed]
- Qu, R.; Ling, Y.; Zhang, Y.-H.-Z.; Wei, L.-Y.; Chen, X.; Li, X.-M.; Liu, X.-Y.; Liu, H.-M.; Guo, Z.; Ren, H.; et al. Platelet-to-Lymphocyte Ratio Is Associated with Prognosis in Patients with Coronavirus Disease-19. J. Med. Virol. 2020, 92, 1533–1541. [Google Scholar] [CrossRef]
- Yang, A.-P.; Liu, J.-P.; Tao, W.-Q.; Li, H.-M. The Diagnostic and Predictive Role of NLR, d-NLR and PLR in COVID-19 Patients. Int. Immunopharmacol. 2020, 84, 106504. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Moore, J.B.; June, C.H. Cytokine Release Syndrome in Severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Levi, M.; Levy, J.H. Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation. Semin. Thromb. Hemost. 2020, 46, 89–95. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef]
- Abou-Ismail, M.Y.; Diamond, A.; Kapoor, S.; Arafah, Y.; Nayak, L. The Hypercoagulable State in COVID-19: Incidence, Pathophysiology, and Management. Thromb. Res. 2020, 194, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological Findings of COVID-19 Associated with Acute Respiratory Distress Syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kanneganti, T.-D. Innate Immunity: The First Line of Defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, S.; Nouri, Z.; Moradi, S.Z.; Akkol, E.K.; Piri, S.; Sobarzo-Sánchez, E.; Farzaei, M.H.; Echeverría, J. Targeting Multiple Signal Transduction Pathways of SARS-CoV-2: Approaches to COVID-19 Therapeutic Candidates. Molecules 2021, 26, 2917. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 Infects Cells after Viral Entry via Clathrin-Mediated Endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Channappanavar, R.; Kanneganti, T.-D. Coronaviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Trends Immunol. 2020, 41, 1083–1099. [Google Scholar] [CrossRef]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The Signal Pathways and Treatment of Cytokine Storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255. [Google Scholar] [CrossRef]
- Magro, G. SARS-CoV-2 and COVID-19: Is Interleukin-6 (IL-6) the “culprit Lesion” of ARDS Onset? What Is There besides Tocilizumab? SGP130Fc. Cytokine X 2020, 2, 100029. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L.; Alphonse, N.; Dickenson, R.E.; Durbin, J.E.; Glenn, J.S.; Hartmann, R.; Kotenko, S.V.; Lazear, H.M.; O’Brien, T.R.; Odendall, C.; et al. COVID-19 and Emerging Viral Infections: The Case for Interferon Lambda. J. Exp. Med. 2020, 217, e20200653. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, M.; Maini, R.N.; Woody, J.N.; Holgate, S.T.; Winter, G.; Rowland, M.; Richards, D.; Hussell, T. Trials of Anti-Tumour Necrosis Factor Therapy for COVID-19 Are Urgently Needed. Lancet 2020, 395, 1407–1409. [Google Scholar] [CrossRef]
- Angelopoulou, A.; Alexandris, N.; Konstantinou, E.; Mesiakaris, K.; Zanidis, C.; Farsalinos, K.; Poulas, K. Imiquimod—A Toll like Receptor 7 Agonist—Is an Ideal Option for Management of COVID 19. Environ. Res. 2020, 188, 109858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wu, Z.; Li, J.-W.; Zhao, H.; Wang, G.-Q. Cytokine Release Syndrome in Severe COVID-19: Interleukin-6 Receptor Antagonist Tocilizumab May Be the Key to Reduce Mortality. Int. J. Antimicrob. Agents 2020, 55, 105954. [Google Scholar] [CrossRef]
- Goker Bagca, B.; Biray Avci, C. The Potential of JAK/STAT Pathway Inhibition by Ruxolitinib in the Treatment of COVID-19. Cytokine Growth Factor Rev. 2020, 54, 51–61. [Google Scholar] [CrossRef]
- Choudhary, S.; Sharma, K.; Silakari, O. The Interplay between Inflammatory Pathways and COVID-19: A Critical Review on Pathogenesis and Therapeutic Options. Microb. Pathog. 2021, 150, 104673. [Google Scholar] [CrossRef] [PubMed]
- Hussman, J.P. Cellular and Molecular Pathways of COVID-19 and Potential Points of Therapeutic Intervention. Front. Pharmacol. 2020, 11, 1169. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Bronte, V.; Ugel, S.; Tinazzi, E.; Vella, A.; Sanctis, F.D.; Canè, S.; Batani, V.; Trovato, R.; Fiore, A.; Petrova, V.; et al. Baricitinib Restrains the Immune Dysregulation in Patients with Severe COVID-19. J. Clin. Investig. 2020, 130, 6409–6416. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Satarker, S.; Tom, A.A.; Shaji, R.A.; Alosious, A.; Luvis, M.; Nampoothiri, M. JAK-STAT Pathway Inhibition and Their Implications in COVID-19 Therapy. Postgrad. Med. 2021, 133, 489–507. [Google Scholar] [CrossRef] [PubMed]
- Seif, F.; Aazami, H.; Khoshmirsafa, M.; Kamali, M.; Mohsenzadegan, M.; Pornour, M.; Mansouri, D. JAK Inhibition as a New Treatment Strategy for Patients with COVID-19. Int. Arch. Allergy Immunol. 2020, 181, 467–475. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, M.; Ghosh, M.K. Hemophagoctic Lymphohistiocytosis—Recent Concept. J. Assoc. Physicians India 2008, 56, 453–457. [Google Scholar] [PubMed]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.H.; Webster, R.G. Tumor Necrosis Factor Alpha Exerts Powerful Anti-Influenza Virus Effects in Lung Epithelial Cells. J. Virol. 2002, 76, 1071–1076. [Google Scholar] [CrossRef] [Green Version]
- Cheung, C.Y.; Poon, L.L.M.; Ng, I.H.Y.; Luk, W.; Sia, S.-F.; Wu, M.H.S.; Chan, K.-H.; Yuen, K.-Y.; Gordon, S.; Guan, Y.; et al. Cytokine Responses in Severe Acute Respiratory Syndrome Coronavirus-Infected Macrophages In Vitro: Possible Relevance to Pathogenesis. J. Virol. 2005, 79, 7819–7826. [Google Scholar] [CrossRef] [Green Version]
- Law, H.K.W.; Cheung, C.Y.; Ng, H.Y.; Sia, S.F.; Chan, Y.O.; Luk, W.; Nicholls, J.M.; Peiris, J.S.M.; Lau, Y.L. Chemokine Up-Regulation in SARS-Coronavirus–Infected, Monocyte-Derived Human Dendritic Cells. Blood 2005, 106, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Mahallawi, W.H.; Khabour, O.F.; Zhang, Q.; Makhdoum, H.M.; Suliman, B.A. MERS-CoV Infection in Humans Is Associated with a pro-Inflammatory Th1 and Th17 Cytokine Profile. Cytokine 2018, 104, 8–13. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-ΚB-Mediated Inflammation in Severe Acute Respiratory Syndrome Coronavirus-Infected Mice Increases Survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef]
- Wan, S.; Yi, Q.; Fan, S.; Lv, J.; Zhang, X.; Guo, L.; Lang, C.; Xiao, Q.; Xiao, K.; Yi, Z.; et al. Characteristics of Lymphocyte Subsets and Cytokines in Peripheral Blood of 123 Hospitalized Patients with 2019 Novel Coronavirus Pneumonia (NCP). medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Komaravelli, N.; Casola, A. Respiratory Viral Infections and Subversion of Cellular Antioxidant Defenses. J. Pharm. Pharm. 2014, 5, 1000141. [Google Scholar] [CrossRef]
- Delgado-Roche, L.; Mesta, F. Oxidative Stress as Key Player in Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Infection. Arch. Med. Res. 2020, 51, 384–387. [Google Scholar] [CrossRef]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune Response in COVID-19: Addressing a Pharmacological Challenge by Targeting Pathways Triggered by SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 84. [Google Scholar] [CrossRef]
- Pétrilli, V.; Dostert, C.; Muruve, D.A.; Tschopp, J. The Inflammasome: A Danger Sensing Complex Triggering Innate Immunity. Curr. Opin. Immunol. 2007, 19, 615–622. [Google Scholar] [CrossRef]
- He, L.; Ding, Y.; Zhang, Q.; Che, X.; He, Y.; Shen, H.; Wang, H.; Li, Z.; Zhao, L.; Geng, J.; et al. Expression of Elevated Levels of Pro-Inflammatory Cytokines in SARS-CoV-Infected ACE2+ Cells in SARS Patients: Relation to the Acute Lung Injury and Pathogenesis of SARS. J. Pathol. 2006, 210, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Alosaimi, B.; Hamed, M.E.; Naeem, A.; Alsharef, A.A.; AlQahtani, S.Y.; AlDosari, K.M.; Alamri, A.A.; Al-Eisa, K.; Khojah, T.; Assiri, A.M.; et al. MERS-CoV Infection Is Associated with Downregulation of Genes Encoding Th1 and Th2 Cytokines/Chemokines and Elevated Inflammatory Innate Immune Response in the Lower Respiratory Tract. Cytokine 2020, 126, 154895. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Lau, C.C.Y.; Chan, K.-H.; Li, C.P.Y.; Chen, H.; Jin, D.-Y.; Chan, J.F.W.; Woo, P.C.Y.; Yuen, K.-Y. 2013 Delayed Induction of Proinflammatory Cytokines and Suppression of Innate Antiviral Response by the Novel Middle East Respiratory Syndrome Coronavirus: Implications for Pathogenesis and Treatment. J. Gen. Virol. 2013, 94, 2679–2690. [Google Scholar] [CrossRef]
- Shi, C.-S.; Nabar, N.R.; Huang, N.-N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b Triggers Intracellular Stress Pathways and Activates NLRP3 Inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavizadeh, L.; Ghasemi, S. Genotype and Phenotype of COVID-19: Their Roles in Pathogenesis. J. Microbiol. Immunol. Infect. 2021, 54, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.M.; Persechini, P.M.; Ojcius, D.M. ATP Activates a Reactive Oxygen Species-Dependent Oxidative Stress Response and Secretion of Proinflammatory Cytokines in Macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.K.; Wu, H.; Yan, H.; Ma, S.; Wang, L.; Zhang, M.; Tang, X.; Temperton, N.J.; Weiss, R.A.; Brenchley, J.M.; et al. T Cell Responses to Whole SARS Coronavirus in Humans. J. Immunol. 2008, 181, 5490–5500. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Fett, C.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Virus-Specific Memory CD8 T Cells Provide Substantial Protection from Lethal Severe Acute Respiratory Syndrome Coronavirus Infection. J. Virol. 2014, 88, 11034–11044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Wang, W.; Yin, J.; Ouyang, Y.; Pang, L.; Feng, Y.; Qiao, L.; Guo, X.; Shi, H.; Jin, R.; et al. The Inhibition of IL-2/IL-2R Gives Rise to CD8+ T Cell and Lymphocyte Decrease through JAK1-STAT5 in Critical Patients with COVID-19 Pneumonia. Cell Death Dis. 2020, 11, 429. [Google Scholar] [CrossRef] [PubMed]
- Bekele, Y.; Sui, Y.; Berzofsky, J.A. IL-7 in SARS-CoV-2 Infection and as a Potential Vaccine Adjuvant. Front. Immunol. 2021, 12, 3796. [Google Scholar] [CrossRef]
- Park, J.-H.; Yu, Q.; Erman, B.; Appelbaum, J.S.; Montoya-Durango, D.; Grimes, H.L.; Singer, A. Suppression of IL7Ralpha Transcription by IL-7 and Other Prosurvival Cytokines: A Novel Mechanism for Maximizing IL-7-Dependent T Cell Survival. Immunity 2004, 21, 289–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, M.; O’Garra, A. The Regulation of IL-10 Production by Immune Cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, J.M.; Avia, M.; Martín, V.; Sevilla, N. IL-10: A Multifunctional Cytokine in Viral Infections. J. Immunol. Res. 2017, 2017, 6104054. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling Serum Cytokines in COVID-19 Patients Reveals IL-6 and IL-10 Are Disease Severity Predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Mal, X.; Trinchieri, G. Regulation of Interleukin-12 Production in Antigen-Presenting Cells. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2001; Volume 79, pp. 55–92. [Google Scholar]
- O’Shea, J.J.; Paul, W.E. Regulation of THI Differentiation—Controlling the Controllers. Nat. Immunol. 2002, 3, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Vecchié, A.; Wang, T.S.; Lee, E.; Cremer, P.C.; Carey, B.; Rajendram, P.; Hudock, K.M.; Korbee, L.; Van Tassell, B.W.; et al. Targeting GM-CSF in COVID-19 Pneumonia: Rationale and Strategies. Front. Immunol. 2020, 11, 1625. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Plebani, M. Laboratory Abnormalities in Patients with COVID-2019 Infection. Clin. Chem. Lab. Med. 2020, 58, 1131–1134. [Google Scholar] [CrossRef] [Green Version]
- Fathi, N.; Rezaei, N. Lymphopenia in COVID-19: Therapeutic Opportunities. Cell Biol. Int. 2020, 44, 1792–1797. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and Biochemical Indexes from 2019-NCoV Infected Patients Linked to Viral Loads and Lung Injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.-Q.; Wang, Q.; Miao, H. Lymphopenia Predicts Disease Severity of COVID-19: A Descriptive and Predictive Study. Signal Transduct. Target. Ther. 2020, 5, 33. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Cameron, M.J.; Ran, L.; Xu, L.; Danesh, A.; Bermejo-Martin, J.F.; Cameron, C.M.; Muller, M.P.; Gold, W.L.; Richardson, S.E.; Poutanen, S.M.; et al. Interferon-Mediated Immunopathological Events Are Associated with Atypical Innate and Adaptive Immune Responses in Patients with Severe Acute Respiratory Syndrome. J. Virol. 2007, 81, 8692–8706. [Google Scholar] [CrossRef] [Green Version]
- Fung, S.-Y.; Yuen, K.-S.; Ye, Z.-W.; Chan, C.-P.; Jin, D.-Y. A Tug-of-War between Severe Acute Respiratory Syndrome Coronavirus 2 and Host Antiviral Defence: Lessons from Other Pathogenic Viruses. Emerg. Microbes Infect. 2020, 9, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xie, J.; Zhao, L.; Fei, X.; Zhang, H.; Tan, Y.; Nie, X.; Zhou, L.; Liu, Z.; Ren, Y.; et al. Alveolar Macrophage Dysfunction and Cytokine Storm in the Pathogenesis of Two Severe COVID-19 Patients. EBioMedicine 2020, 57, 102833. [Google Scholar] [CrossRef] [PubMed]
- Al-Saadi, E.A.K.D.; Abdulnabi, M.A. Hematological Changes Associated with COVID-19 Infection. J. Clin. Lab. Anal. 2022, 36, e24064. [Google Scholar] [CrossRef] [PubMed]
- Pasrija, R.; Naime, M. Resolving the Equation between Mucormycosis and COVID-19 Disease. Mol. Biol. Rep. 2022, 49, 3349–3356. [Google Scholar] [CrossRef] [PubMed]
- Menéndez, R.; Méndez, R.; Almansa, R.; Ortega, A.; Alonso, R.; Suescun, M.; Ferrando, A.; Feced, L.; Bermejo-Martin, J.F. Simultaneous Depression of Immunological Synapse and Endothelial Injury Is Associated with Organ Dysfunction in Community-Acquired Pneumonia. J. Clin. Med. 2019, 8, 1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Henry, B.M.; de Oliveira, M.H.S.; Benoit, S.; Plebani, M.; Lippi, G. Hematologic, Biochemical and Immune Biomarker Abnormalities Associated with Severe Illness and Mortality in Coronavirus Disease 2019 (COVID-19): A Meta-Analysis. Clin. Chem. Lab. Med. 2020, 58, 1021–1028. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Wang, Y.; Liang, X.; Xiao, W.; Duan, G.; Yang, H.; Wang, Y. Is Neutrophilia Associated with Mortality in COVID-19 Patients? A Meta-Analysis and Meta-Regression. Int. J. Lab. Hematol. 2020, 42, e244–e247. [Google Scholar] [CrossRef]
- Sun, S.; Cai, X.; Wang, H.; He, G.; Lin, Y.; Lu, B.; Chen, C.; Pan, Y.; Hu, X. Abnormalities of Peripheral Blood System in Patients with COVID-19 in Wenzhou, China. Clin. Chim. Acta 2020, 507, 174–180. [Google Scholar] [CrossRef]
- Kong, J.; Wang, T.; Di, Z.; Shi, B.; Yu, X.; Huang, C.; Yang, Y.; Sun, H.; Yuan, D.; Wu, D.; et al. Analysis of Hematological Indexes of COVID-19 Patients from Fever Clinics in Suzhou, China. Int. J. Lab. Hematol. 2020, 42, e204–e206. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, X.; Kong, M.; Mao, X.; Huang, L.; He, P.; Pan, S.; Li, J.; Lu, Z. Clinical and Hematological Characteristics of 88 Patients with COVID-19. Int. J. Lab. Hematol. 2020, 42, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, X.; Chen, J.; Jin, Y.; Peng, L.; Wang, H.H.X.; Luo, M.; Chen, L.; Zhao, Y. Neutrophil-to-Lymphocyte Ratio as an Independent Risk Factor for Mortality in Hospitalized Patients with COVID-19. J. Infect. 2020, 81, e6–e12. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological Findings and Complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T-Cells and Inflammatory Monocytes Incite Inflammatory Storms in Severe COVID-19 Patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Guo, R.; Lei, L.; Liu, H.; Wang, Y.; Wang, Y.; Qian, H.; Dai, T.; Zhang, T.; Lai, Y.; et al. COVID-19 Infection Induces Readily Detectable Morphologic and Inflammation-related Phenotypic Changes in Peripheral Blood Monocytes. J. Leukoc. Biol. 2020, 109, 13–22. [Google Scholar] [CrossRef]
- Gómez-Rial, J.; Currás-Tuala, M.J.; Rivero-Calle, I.; Gómez-Carballa, A.; Cebey-López, M.; Rodríguez-Tenreiro, C.; Dacosta-Urbieta, A.; Rivero-Velasco, C.; Rodríguez-Núñez, N.; Trastoy-Pena, R.; et al. Increased Serum Levels of SCD14 and SCD163 Indicate a Preponderant Role for Monocytes in COVID-19 Immunopathology. Front. Immunol. 2020, 11, 560381. [Google Scholar] [CrossRef]
- Pence, B.D. Severe COVID-19 and Aging: Are Monocytes the Key? GeroScience 2020, 42, 1051–1061. [Google Scholar] [CrossRef]
- Hu, B.; Huang, S.; Yin, L. The Cytokine Storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 Patients in Intensive Care Unit: A Report of Thromboelastography Findings and Other Parameters of Hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef]
- Rokni, M.; Hamblin, M.R.; Rezaei, N. Cytokines and COVID-19: Friends or Foes? Hum. Vaccines Immunother. 2020, 16, 2363–2365. [Google Scholar] [CrossRef]
- Knoll, R.; Schultze, J.L.; Schulte-Schrepping, J. Monocytes and Macrophages in COVID-19. Front. Immunol. 2021, 12, 2952. [Google Scholar] [CrossRef] [PubMed]
- Yilla, M.; Harcourt, B.H.; Hickman, C.J.; McGrew, M.; Tamin, A.; Goldsmith, C.S.; Bellini, W.J.; Anderson, L.J. SARS-Coronavirus Replication in Human Peripheral Monocytes/Macrophages. Virus Res. 2005, 107, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.S.; Bentz, G.L.; Alexander, J.S.; Yurochko, A.D. Human Cytomegalovirus Induces Monocyte Differentiation and Migration as a Strategy for Dissemination and Persistence. J. Virol. 2004, 78, 4444–4453. [Google Scholar] [CrossRef] [Green Version]
- Nottet, H.S.; Persidsky, Y.; Sasseville, V.G.; Nukuna, A.N.; Bock, P.; Zhai, Q.H.; Sharer, L.R.; McComb, R.D.; Swindells, S.; Soderland, C.; et al. Mechanisms for the Transendothelial Migration of HIV-1-Infected Monocytes into Brain. J. Immunol. 1996, 156, 1284–1295. [Google Scholar]
- Desforges, M.; Miletti, T.C.; Gagnon, M.; Talbot, P.J. Activation of Human Monocytes after Infection by Human Coronavirus 229E. Virus Res. 2007, 130, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, A.A.; Lyroni, K.; Aznaourova, M.; Tseliou, M.; Al-Anazi, M.R.; Al-Ahdal, M.N.; Alkahtani, S.; Sourvinos, G.; Tsatsanis, C. Middle East Respiratory Syndrome Corona Virus Spike Glycoprotein Suppresses Macrophage Responses via DPP4-Mediated Induction of IRAK-M and PPARγ. Oncotarget 2017, 8, 9053–9066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitina, E.; Larionova, I.; Choinzonov, E.; Kzhyshkowska, J. Monocytes and Macrophages as Viral Targets and Reservoirs. Int. J. Mol. Sci. 2018, 19, 2821. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, S.; Subbian, S. Critical Determinants of Cytokine Storm and Type I Interferon Response in COVID-19 Pathogenesis. Clin. Microbiol. Rev. 2021, 34, e00299-20. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Mossel, E.C.; Sainz, B.; Garry, R.F.; Peters, C.J. Synergistic Inhibition of SARS-Coronavirus Replication by Type I and Type II IFN. Adv. Exp. Med. Biol. 2006, 581, 503–506. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokugamage, K.G.; Hage, A.; de Vries, M.; Valero-Jimenez, A.M.; Schindewolf, C.; Dittmann, M.; Rajsbaum, R.; Menachery, V.D. Type I Interferon Susceptibility Distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 2020, 94, e01410-20. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xu, J.; Zhou, C.; Wu, Z.; Zhong, S.; Liu, J.; Luo, W.; Chen, T.; Qin, Q.; Deng, P. Characterization of Cytokine/Chemokine Profiles of Severe Acute Respiratory Syndrome. Am. J. Respir. Crit. Care Med. 2005, 171, 850–857. [Google Scholar] [CrossRef]
- Sinha, P.; Matthay, M.A.; Calfee, C.S. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern. Med. 2020, 180, 1152–1154. [Google Scholar] [CrossRef]
- Hedrick, T.L.; Murray, B.P.; Hagan, R.S.; Mock, J.R. COVID-19: Clean up on IL-6. Am. J. Respir. Cell Mol. Biol. 2020, 63, 541–543. [Google Scholar] [CrossRef]
- Wiseman, D.; Lin, J.; Routy, J.-P.; Samoukovic, G. Haemophagocytic Lymphohistiocytosis in an Adult with Postacute COVID-19 Syndrome. BMJ Case Rep. 2021, 14, e245031. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Berliner, N. Hemophagocytic Lymphohistiocytosis. Annu. Rev. Pathol. 2018, 13, 27–49. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zerón, P.; López-Guillermo, A.; Khamashta, M.A.; Bosch, X. Adult Haemophagocytic Syndrome. Lancet 2014, 383, 1503–1516. [Google Scholar] [CrossRef]
- Yu, Z.; Ellahi, R.; Nutini, A.; Sohail, A.; Sait, S.M. Modeling and Simulations of CoViD-19 Molecular Mechanism Induced by Cytokines Storm during SARS-CoV2 Infection. J. Mol. Liq. 2021, 327, 114863. [Google Scholar] [CrossRef]
- Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Influenza Infection, SARS, MERS and COVID-19: Cytokine Storm—The Common Denominator and the Lessons to Be Learned. Clin. Immunol. 2021, 223, 108652. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Moochhala, S. Role of Inflammatory Mediators in the Pathophysiology of Acute Respiratory Distress Syndrome. J. Pathol. 2004, 202, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Lan, Z.; Ye, J.; Pang, L.; Liu, Y.; Wu, W.; Qin, X.; Guo, Y.; Zhang, P. Cytokine Storm: The Primary Determinant for the Pathophysiological Evolution of COVID-19 Deterioration. Front. Immunol. 2021, 12, 1409. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, T.; Cai, D.; Hu, Z.; Chen, J.; Liao, H.; Zhi, L.; Wei, H.; Zhang, Z.; Qiu, Y.; et al. Cytokine Storm Intervention in the Early Stages of COVID-19 Pneumonia. Cytokine Growth Factor Rev. 2020, 53, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Moradian, N.; Gouravani, M.; Salehi, M.A.; Heidari, A.; Shafeghat, M.; Hamblin, M.R.; Rezaei, N. Cytokine Release Syndrome: Inhibition of pro-Inflammatory Cytokines as a Solution for Reducing COVID-19 Mortality. Eur. Cytokine Netw. 2020, 31, 81–93. [Google Scholar] [CrossRef]
- Tang, L.; Yin, Z.; Hu, Y.; Mei, H. Controlling Cytokine Storm Is Vital in COVID-19. Front. Immunol. 2020, 11, 570993. [Google Scholar] [CrossRef]
- Liu, T.; Balzano-Nogueira, L.; Lleo, A.; Conesa, A. Transcriptional Differences for COVID-19 Disease Map Genes between Males and Females Indicate a Different Basal Immunophenotype Relevant to the Disease. Genes 2020, 11, 1447. [Google Scholar] [CrossRef]
- Docherty, A.B.; Harrison, E.M.; Green, C.A.; Hardwick, H.E.; Pius, R.; Norman, L.; Holden, K.A.; Read, J.M.; Dondelinger, F.; Carson, G.; et al. Features of 20,133 UK Patients in Hospital with Covid-19 Using the ISARIC WHO Clinical Characterisation Protocol: Prospective Observational Cohort Study. BMJ 2020, 369, m1985. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Jia, X.; Li, J.; Hu, K.; Chen, G.; Wei, J.; Gong, Z.; Zhou, C.; Yu, H.; et al. Risk Factors for Disease Severity, Unimprovement, and Mortality in COVID-19 Patients in Wuhan, China. Clin. Microbiol. Infect. 2020, 26, 767–772. [Google Scholar] [CrossRef]
- Sharma, G.; Volgman, A.S.; Michos, E.D. Sex Differences in Mortality from COVID-19 Pandemic. JACC Case Rep. 2020, 2, 1407–1410. [Google Scholar] [CrossRef]
- Gadi, N.; Wu, S.C.; Spihlman, A.P.; Moulton, V.R. What’s Sex Got to Do with COVID-19? Gender-Based Differences in the Host Immune Response to Coronaviruses. Front. Immunol. 2020, 11, 2147. [Google Scholar] [CrossRef] [PubMed]
- Scully, E.P.; Haverfield, J.; Ursin, R.L.; Tannenbaum, C.; Klein, S.L. Considering How Biological Sex Impacts Immune Responses and COVID-19 Outcomes. Nat. Rev. Immunol. 2020, 20, 442–447. [Google Scholar] [CrossRef]
- Rhodes, J.M.; Subramanian, S.; Laird, E.; Griffin, G.; Kenny, R.A. Perspective: Vitamin D Deficiency and COVID-19 Severity—Plausibly Linked by Latitude, Ethnicity, Impacts on Cytokines, ACE2, and Thrombosis (R1). J. Intern. Med. 2020, 289, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.T.; Peruzzu, D.; Ruggieri, A.; Ortona, E.; Gagliardi, M.C. Vitamin D and Sex Differences in COVID-19. Front. Endocrinol. 2020, 11, 567824. [Google Scholar] [CrossRef] [PubMed]
- Laird, E.; McNulty, H.; Ward, M.; Hoey, L.; McSorley, E.; Wallace, J.M.W.; Carson, E.; Molloy, A.M.; Healy, M.; Casey, M.C.; et al. Vitamin D Deficiency Is Associated with Inflammation in Older Irish Adults. J. Clin. Endocrinol. Metab. 2014, 99, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Parpia, A.S.; Martinez, I.; El-Sayed, A.M.; Wells, C.R.; Myers, L.; Duncan, J.; Collins, J.; Fitzpatrick, M.C.; Galvani, A.P.; Pandey, A. Racial Disparities in COVID-19 Mortality across Michigan, United States. EClinicalMedicine 2021, 33, 100761. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors Associated with COVID-19-Related Death Using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef]
- Golestaneh, L.; Neugarten, J.; Fisher, M.; Billett, H.H.; Gil, M.R.; Johns, T.; Yunes, M.; Mokrzycki, M.H.; Coco, M.; Norris, K.C.; et al. The Association of Race and COVID-19 Mortality. EClinicalMedicine 2020, 25, 100455. [Google Scholar] [CrossRef]
- Maurya, R.; Sebastian, P.; Namdeo, M.; Devender, M.; Gertler, A. COVID-19 Severity in Obesity: Leptin and Inflammatory Cytokine Interplay in the Link Between High Morbidity and Mortality. Front. Immunol. 2021, 12, 2349. [Google Scholar] [CrossRef]
- Gammone, M.A.; D’Orazio, N. Review: Obesity and COVID-19: A Detrimental Intersection. Front. Endocrinol. 2021, 12, 396. [Google Scholar] [CrossRef]
- Kim, J.; Nam, J.-H. Insight into the Relationship between Obesity-Induced Low-Level Chronic Inflammation and COVID-19 Infection. Int. J. Obes. 2020, 44, 1541–1542. [Google Scholar] [CrossRef] [PubMed]
- Leija-Martínez, J.J.; Huang, F.; Del-Río-Navarro, B.E.; Sanchéz-Muñoz, F.; Muñoz-Hernández, O.; Giacoman-Martínez, A.; Hall-Mondragon, M.S.; Espinosa-Velazquez, D. IL-17A and TNF-α as Potential Biomarkers for Acute Respiratory Distress Syndrome and Mortality in Patients with Obesity and COVID-19. Med. Hypotheses 2020, 144, 109935. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Luo, Y.; Huang, C.; Wang, Z.; Song, G.; Pan, Y.; Zhao, X.; Liu, S. An Intelligent Graphene-Based Biosensing Device for Cytokine Storm Syndrome Biomarkers Detection in Human Biofluids. Small 2021, 17, 2101508. [Google Scholar] [CrossRef] [PubMed]
- Strope, J.D.; PharmD, C.H.C.; Figg, W.D. TMPRSS2: Potential Biomarker for COVID-19 Outcomes. J. Clin. Pharmacol. 2020, 60, 801–807. [Google Scholar] [CrossRef]
- Liu, T.; Yi, J.Z.; Yang, Y.; Ma, H.; Li, Z.; Zhang, J.; Cheng, J.; Zhang, X.; Zhao, Y.; Xia, Z.; et al. The Role of Interleukin-6 in Monitoring Severe Case of Coronavirus Disease 2019. EMBO Mol. Med. 2020, 12, e12421. [Google Scholar] [CrossRef]
- Liu, B.M.; Martins, T.B.; Peterson, L.K.; Hill, H.R. Clinical Significance of Measuring Serum Cytokine Levels as Inflammatory Biomarkers in Adult and Pediatric COVID-19 Cases: A Review. Cytokine 2021, 142, 155478. [Google Scholar] [CrossRef]
- Titanji, B.K.; Farley, M.M.; Mehta, A.; Connor-Schuler, R.; Moanna, A.; Cribbs, S.K.; O’Shea, J.; DeSilva, K.; Chan, B.; Edwards, A.; et al. Use of Baricitinib in Patients with Moderate to Severe Coronavirus Disease 2019. Clin. Infect. Dis. 2021, 72, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Cataño-Correa, J.C.; Cardona-Arias, J.A.; Porras Mancilla, J.P.; García, M.T. Bacterial Superinfection in Adults with COVID-19 Hospitalized in Two Clinics in Medellín-Colombia, 2020. PLoS ONE 2021, 16, e0254671. [Google Scholar] [CrossRef] [PubMed]
- Geppert, A.; Steiner, A.; Zorn, G.; Delle-Karth, G.; Koreny, M.; Haumer, M.; Siostrzonek, P.; Huber, K.; Heinz, G. Multiple Organ Failure in Patients with Cardiogenic Shock Is Associated with High Plasma Levels of Interleukin-6. Crit. Care Med. 2002, 30, 1987–1994. [Google Scholar] [CrossRef]
- Melo, A.K.G.; Milby, K.M.; Caparroz, A.L.M.A.; Pinto, A.C.P.N.; Santos, R.R.P.; Rocha, A.P.; Ferreira, G.A.; Souza, V.A.; Valadares, L.D.A.; Vieira, R.M.R.A.; et al. Biomarkers of Cytokine Storm as Red Flags for Severe and Fatal COVID-19 Cases: A Living Systematic Review and Meta-Analysis. PLoS ONE 2021, 16, e0253894. [Google Scholar] [CrossRef]
- Sims, J.T.; Krishnan, V.; Chang, C.-Y.; Engle, S.M.; Casalini, G.; Rodgers, G.H.; Bivi, N.; Nickoloff, B.J.; Konrad, R.J.; de Bono, S.; et al. Characterization of the Cytokine Storm Reflects Hyperinflammatory Endothelial Dysfunction in COVID-19. J. Allergy Clin. Immunol. 2021, 147, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Eljaaly, K.; Malibary, H.; Alsulami, S.; Albanji, M.; Badawi, M.; Al-Tawfiq, J.A. Description and Analysis of Cytokine Storm in Registered COVID-19 Clinical Trials: A Systematic Review. Pathogens 2021, 10, 692. [Google Scholar] [CrossRef]
- Stockman, L.J.; Bellamy, R.; Garner, P. SARS: Systematic Review of Treatment Effects. PLoS Med. 2006, 3, e343. [Google Scholar] [CrossRef] [Green Version]
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral Activities of Type I Interferons to SARS-CoV-2 Infection. Antivir. Res. 2020, 179, 104811. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, V.; Shannon, C.P.; Wei, X.-S.; Xiang, X.; Wang, X.; Wang, Z.-H.; Tebbutt, S.J.; Kollmann, T.R.; Fish, E.N. Interferon-A2b Treatment for COVID-19. Front. Immunol. 2020, 11, 1061. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Wang, T.; Chen, L.; Chen, X.; Li, L.; Qin, X.; Li, H.; Luo, J. The Effect of Recombinant Human Interferon Alpha Nasal Drops to Prevent COVID-19 Pneumonia for Medical Staff in an Epidemic Area. Curr. Top. Med. Chem. 2021, 21, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L.; Hou, Y.J.; Adams, L.E.; et al. Publisher Correction: A Mouse-Adapted Model of SARS-CoV-2 to Test COVID-19 Countermeasures. Nature 2021, 590, E22. [Google Scholar] [CrossRef] [PubMed]
- Monneret, G.; de Marignan, D.; Coudereau, R.; Bernet, C.; Ader, F.; Frobert, E.; Gossez, M.; Viel, S.; Venet, F.; Wallet, F. Immune Monitoring of Interleukin-7 Compassionate Use in a Critically Ill COVID-19 Patient. Cell. Mol. Immunol. 2020, 17, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A. Background to New Treatments for COVID-19, Including Its Chronicity, through Altering Elements of the Cytokine Storm. Rev. Med. Virol. 2021, 31, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sebba, A. Tocilizumab: The First Interleukin-6-Receptor Inhibitor. Am. J. Health-Syst. Pharm. 2008, 65, 1413–1418. [Google Scholar] [CrossRef]
- Tomasiewicz, K.; Piekarska, A.; Stempkowska-Rejek, J.; Serafińska, S.; Gawkowska, A.; Parczewski, M.; Niścigorska-Olsen, J.; Łapiński, T.W.; Zarębska-Michaluk, D.; Kowalska, J.D.; et al. Tocilizumab for Patients with Severe COVID-19: A Retrospective, Multi-Center Study. Expert Rev. Anti-Infect. Ther. 2021, 19, 93–100. [Google Scholar] [CrossRef]
- Choy, E.H.; De Benedetti, F.; Takeuchi, T.; Hashizume, M.; John, M.R.; Kishimoto, T. Translating IL-6 Biology into Effective Treatments. Nat. Rev. Rheumatol. 2020, 16, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Toniati, P.; Piva, S.; Cattalini, M.; Garrafa, E.; Regola, F.; Castelli, F.; Franceschini, F.; Airò, P.; Bazzani, C.; Beindorf, E.-A.; et al. Tocilizumab for the Treatment of Severe COVID-19 Pneumonia with Hyperinflammatory Syndrome and Acute Respiratory Failure: A Single Center Study of 100 Patients in Brescia, Italy. Autoimmun. Rev. 2020, 19, 102568. [Google Scholar] [CrossRef] [PubMed]
- Morena, V.; Milazzo, L.; Oreni, L.; Bestetti, G.; Fossali, T.; Bassoli, C.; Torre, A.; Cossu, M.V.; Minari, C.; Ballone, E.; et al. Off-Label Use of Tocilizumab for the Treatment of SARS-CoV-2 Pneumonia in Milan, Italy. Eur. J. Intern. Med. 2020, 76, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, M.; Mannam, P.; Comer, R.; Sinclair, E.; McQuaid, D.B.; Schmidt, M.L. Off-Label Real World Experience Using Tocilizumab for Patients Hospitalized with COVID-19 Disease in a Regional Community Health System: A Case-Control Study. medRxiv 2020. [Google Scholar] [CrossRef]
- Luo, P.; Liu, Y.; Qiu, L.; Liu, X.; Liu, D.; Li, J. Tocilizumab Treatment in COVID-19: A Single Center Experience. J. Med. Virol. 2020, 92, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective Treatment of Severe COVID-19 Patients with Tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef] [PubMed]
- Villatoro Santos, C.R.; Bhargava, A.; Coyle, M.; Szpunar, S.; Saravolatz, L.D. Tocilizumab: A Retrospective Multi-Center Cohort Study of Critically Ill Patients with COVID-19. Int. J. Clin. Pharmacol. Ther. 2021, 59, 705–712. [Google Scholar] [CrossRef]
- Montesarchio, V.; Parrella, R.; Iommelli, C.; Bianco, A.; Manzillo, E.; Fraganza, F.; Palumbo, C.; Rea, G.; Murino, P.; De Rosa, R.; et al. Outcomes and Biomarker Analyses among Patients with COVID-19 Treated with Interleukin 6 (IL-6) Receptor Antagonist Sarilumab at a Single Institution in Italy. J. Immunother. Cancer 2020, 8, e001089. [Google Scholar] [CrossRef]
- Gritti, G.; Raimondi, F.; Ripamonti, D.; Riva, I.; Landi, F.; Alborghetti, L.; Frigeni, M.; Damiani, M.; Micò, C.; Fagiuoli, S.; et al. IL-6 Signalling Pathway Inactivation with Siltuximab in Patients with COVID-19 Respiratory Failure: An Observational Cohort Study. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Pasin, L.; Cavalli, G.; Navalesi, P.; Sella, N.; Landoni, G.; Yavorovskiy, A.G.; Likhvantsev, V.V.; Zangrillo, A.; Dagna, L.; Monti, G. Anakinra for Patients with COVID-19: A Meta-Analysis of Non-Randomized Cohort Studies. Eur. J. Intern. Med. 2021, 86, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Ucciferri, C.; Auricchio, A.; Nicola, M.D.; Potere, N.; Abbate, A.; Cipollone, F.; Vecchiet, J.; Falasca, K. Canakinumab in a Subgroup of Patients with COVID-19. Lancet Rheumatol. 2020, 2, e457–e458. [Google Scholar] [CrossRef]
- Duret, P.-M.; Sebbag, E.; Mallick, A.; Gravier, S.; Spielmann, L.; Messer, L. Recovery from COVID-19 in a Patient with Spondyloarthritis Treated with TNF-Alpha Inhibitor Etanercept. Ann. Rheum. Dis. 2020, 79, 1251–1252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, X.; Ni, L.; Di, X.; Ma, B.; Niu, S.; Liu, C.; Reiter, R.J. COVID-19: Melatonin as a Potential Adjuvant Treatment. Life Sci. 2020, 250, 117583. [Google Scholar] [CrossRef] [PubMed]
- Hasan, Z.T.; Atrakji, D.M.Q.Y.M.A.A.; Mehuaiden, D.A.K. The Effect of Melatonin on Thrombosis, Sepsis and Mortality Rate in COVID-19 Patients. Int. J. Infect. Dis. Off. Publ. Int. Soc. Infect. Dis. 2022, 114, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.A.; Heydari, K.; Mehravaran, H.; Saeedi, M.; Alizadeh-Navaei, R.; Hedayatizadeh-Omran, A.; Shamshirian, A. Melatonin Effects on Sleep Quality and Outcomes of COVID-19 Patients: An Open-label, Randomized, Controlled Trial. J. Med. Virol. 2022, 94, 263–271. [Google Scholar] [CrossRef]
- Bai, F.; Li, G.G.; Liu, Q.; Niu, X.; Li, R.; Ma, H. Short-Term Efficacy and Safety of IL-17, IL-12/23, and IL-23 Inhibitors Brodalumab, Secukinumab, Ixekizumab, Ustekinumab, Guselkumab, Tildrakizumab, and Risankizumab for the Treatment of Moderate to Severe Plaque Psoriasis: A Systematic Review and Network Meta-Analysis of Randomized Controlled Trials. J. Immunol. Res. 2019, 2019, 2546161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almradi, A.; Hanzel, J.; Sedano, R.; Parker, C.E.; Feagan, B.G.; Ma, C.; Jairath, V. Clinical Trials of IL-12/IL-23 Inhibitors in Inflammatory Bowel Disease. BioDrugs 2020, 34, 713–721. [Google Scholar] [CrossRef]
- Ward, M.; Gooderham, M. Asymptomatic SARS-CoV2 Infection in a Patient Receiving Risankizumab, an Inhibitor of Interleukin 23. JAAD Case Rep. 2021, 7, 60–61. [Google Scholar] [CrossRef] [PubMed]
- Kiss, N.; Lorincz, K.; Medvecz, M.; Fésűs, L.; Csuha, P.; Hermányi, Z.; Wikonkal, N. Coronavirus Disease 2019 in a Psoriatic Patient with Concomitant Chronic Obstructive Pulmonary Disease under Treatment with Risankizumab. Dermatol. Ther. 2020, 33, e14186. [Google Scholar] [CrossRef] [PubMed]
- Messina, F.; Piaserico, S. SARS-CoV-2 Infection in a Psoriatic Patient Treated with IL-23 Inhibitor. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e254–e255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messina, F.; Pampaloni, F.; Piaserico, S. Correspondence on ‘Recovery from COVID-19 in a Patient with Spondyloarthritis Treated with TNF-Alpha Inhibitor Etanercept. A Report on a Patient with COVID-19 with Psoriatic Arthritis Receiving Ustekinumab’. Ann. Rheum. Dis. 2020, 80, e79. [Google Scholar] [CrossRef] [PubMed]
- Balestri, R.; Rech, G.; Girardelli, C.R. SARS-CoV-2 Infection in a Psoriatic Patient Treated with IL-17 Inhibitor. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e357–e358. [Google Scholar] [CrossRef]
- Carugno, A.; Gambini, D.M.; Raponi, F.; Vezzoli, P.; Robustelli Test, E.; Arosio, M.E.G.; Callegaro, A.; Sena, P. Coronavirus Disease 2019 (COVID-19) Rash in a Psoriatic Patient Treated with Secukinumab: Is There a Role for Interleukin 17? Dermatol. Ther. 2020, 33, e14011. [Google Scholar] [CrossRef] [PubMed]
- Luca, G.D.; Cavalli, G.; Campochiaro, C.; Della-Torre, E.; Angelillo, P.; Tomelleri, A.; Boffini, N.; Tentori, S.; Mette, F.; Farina, N.; et al. GM-CSF Blockade with Mavrilimumab in Severe COVID-19 Pneumonia and Systemic Hyperinflammation: A Single-Centre, Prospective Cohort Study. Lancet Rheumatol. 2020, 2, e465–e473. [Google Scholar] [CrossRef]
- Temesgen, Z.; Assi, M.; Shweta, F.N.U. GM-CSF Neutralization with Lenzilumab in Severe COVID-19 Pneumonia A Case-Cohort Study. Mayo Clin. Proc. 2020, 95, 2382–2394. [Google Scholar] [CrossRef] [PubMed]
- McLornan, D.P.; Pope, J.E.; Gotlib, J.; Harrison, C.N. Current and Future Status of JAK Inhibitors. Lancet 2021, 398, 803–816. [Google Scholar] [CrossRef]
- Stebbing, J.; Krishnan, V.; de Bono, S.; Ottaviani, S.; Casalini, G.; Richardson, P.J.; Monteil, V.; Lauschke, V.M.; Mirazimi, A.; Youhanna, S.; et al. Mechanism of Baricitinib Supports Artificial Intelligence-Predicted Testing in COVID-19 Patients. EMBO Mol. Med. 2020, 12, e12697. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Rawling, M.; Savory, E.; Stebbing, J. Baricitinib as Potential Treatment for 2019-NCoV Acute Respiratory Disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef] [Green Version]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R.; et al. Efficacy and Safety of Baricitinib for the Treatment of Hospitalised Adults with COVID-19 (COV-BARRIER): A Randomised, Double-Blind, Parallel-Group, Placebo-Controlled Phase 3 Trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Hospitalized Adults: Therapeutic Management. Available online: https://www.covid19treatmentguidelines.nih.gov/management/clinical-management/hospitalized-adults--therapeutic-management/ (accessed on 10 February 2022).
- Cao, Y.; Wei, J.; Zou, L.; Jiang, T.; Wang, G.; Chen, L.; Huang, L.; Meng, F.; Huang, L.; Wang, N.; et al. Ruxolitinib in Treatment of Severe Coronavirus Disease 2019 (COVID-19): A Multicenter, Single-Blind, Randomized Controlled Trial. J. Allergy Clin. Immunol. 2020, 146, 137–146.e3. [Google Scholar] [CrossRef] [PubMed]
- D’Acquisto, F.; May, M.J.; Ghosh, S. Inhibition of Nuclear Factor Kappa B (NF-B). Mol. Interv. 2002, 2, 22. [Google Scholar] [CrossRef]
- Liang, N.; Zhong, Y.; Zhou, J.; Liu, B.; Lu, R.; Guan, Y.; Wang, Q.; Liang, C.; He, Y.; Zhou, Y.; et al. Immunosuppressive Effects of Hydroxychloroquine and Artemisinin Combination Therapy via the Nuclear Factor-κB Signaling Pathway in Lupus Nephritis Mice. Exp. Ther. Med. 2018, 15, 2436–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quagliariello, V.; Bonelli, A.; Caronna, A.; Lombari, M.C.; Conforti, G.; Libutti, M.; Iaffaioli, R.V.; Berretta, M.; Botti, G.; Maurea, N. SARS-CoV-2 Infection: NLRP3 Inflammasome as Plausible Target to Prevent Cardiopulmonary Complications? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9169–9171. [Google Scholar] [PubMed]
- Toldo, S.; Abbate, A. The NLRP3 Inflammasome in Acute Myocardial Infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niedźwiedzka-Rystwej, P.; Majchrzak, A.; Kurkowska, S.; Małkowska, P.; Sierawska, O.; Hrynkiewicz, R.; Parczewski, M. Immune Signature of COVID-19: In-Depth Reasons and Consequences of the Cytokine Storm. Int. J. Mol. Sci. 2022, 23, 4545. https://doi.org/10.3390/ijms23094545
Niedźwiedzka-Rystwej P, Majchrzak A, Kurkowska S, Małkowska P, Sierawska O, Hrynkiewicz R, Parczewski M. Immune Signature of COVID-19: In-Depth Reasons and Consequences of the Cytokine Storm. International Journal of Molecular Sciences. 2022; 23(9):4545. https://doi.org/10.3390/ijms23094545
Chicago/Turabian StyleNiedźwiedzka-Rystwej, Paulina, Adam Majchrzak, Sara Kurkowska, Paulina Małkowska, Olga Sierawska, Rafał Hrynkiewicz, and Miłosz Parczewski. 2022. "Immune Signature of COVID-19: In-Depth Reasons and Consequences of the Cytokine Storm" International Journal of Molecular Sciences 23, no. 9: 4545. https://doi.org/10.3390/ijms23094545