
Abstract
The clinical manifestations of the cytokine storm (CS) associated with COVID-19 resemble the acute phase of sepsis. Metabolomics may contribute to understanding the specific pathobiology of these two syndromes. The aim of this study was to compare serum metabolomic profiles in CS associated with COVID-19 vs. septic surgery patients. In a retrospective cross-sectional study, serum samples from patients with CS associated with COVID-19, with and without comorbidity, as well as serum samples from patients with surgical sepsis were investigated. Targeted metabolomic analysis was performed on all samples using LC–MS/MS. Analysis revealed that similar alterations in the serum metabolome of patients with COVID-19 and surgical septic patients were associated with amino acid metabolism, nitrogen metabolism, inflammatory status, methionine cycle and glycolysis. The most significant difference was found for serum levels of metabolites of kynurenine synthesis, tricarboxylic acid cycle, gamma-aminobutyric acid and niacinamide. The metabolic pathway of cysteine and methionine metabolism was significantly disturbed in COVID-19 and surgical septic patients. For the first time, the similarities and differences between the serum metabolomic profiles of patients with CS associated with COVID-19 and patients with surgical sepsis were investigated for patients from the Northwest of the Russian Federation.
Similar content being viewed by others


Introduction
As of January 14, 2024, the past SARS-CoV-2 pandemic has claimed more than 7 million lives1. Despite a significant number of publications on COVID-19 (391,381 in PubMed as of January 29, 2024), many questions related to the pathophysiology of the disease remain unresolved.
Cytokine storm (CS), observed in a substantial number of COVID-19 patients is extensively discussed syndrome linked to the illness. CS, a systemic inflammatory state characterized by immune cell hyperactivation and uncontrolled cytokine release, is not exclusive to COVID-19. It is known to be triggered by various factors such as infections, tumor processes, autoimmune conditions and others2. CS can cause acute respiratory distress syndrome (ARDS) or multiple organ dysfunction, which can be potentially fatal3.
The clinical manifestations of COVID-19-associated CS and its consequences are similar to those of the acute phase of sepsis4. According to the Third International Consensus Definitions Task Force (Sepsis-3) sepsis is a life-threatening organ dysfunction caused by dysregulation of the host response to infection5. Traditionally, bacterial infection has been considered the major cause of sepsis6. The COVID-19 pandemic has led to a reassessment of the role of viruses in the occurrence of sepsis, because the multi-organ dysfunction caused by CS in COVID-19 largely corresponds to the concept of Sepsis-3 and is currently considered as viral sepsis7.
Sepsis caused by CS associated with COVID-19 exhibits distinctive characteristics, even though its clinical symptoms typically resemble those of bacterial sepsis. COVID-19 is distinguished by a less pronounced and more prolonged occurrence of systemic multi-organ inflammation8,9, an accelerated onset of acute respiratory distress syndrome (ARDS), reduced levels of inflammatory markers like IL-6 in comparison with sepsis10, and an immune signature due to differences in response to bacterial and viral infection4.
Currently, metabolomic profiling of human biological fluids is of great interest because of its potential to provide additional insight into disease pathogenesis and potential therapeutic targets. To date, many studies have been published investigating the metabolomic profile of patients with sepsis8,11,12,13. The pandemic of COVID-19 raised the interest to the metabolomics of this disease and of the CS as its hyperinflammation stage. Thus, since the beginning of the pandemic, numerous studies on this topic have been published14,15,16. Both series of works found the similar alterations in the metabolites associated with energetic of the cells and inflammation as in the amino acids levels both glucogenic and ketogenic. At the same time, there is a lack of studies comparing the metabolomics of these syndromes.
This study focuses on comparing targeted metabolic profiles in the blood serum of patients with surgical sepsis (SS) and those with CS associated with COVID-19. The clinical data of patients with CS do not fully meet the Sepsis-3 criteria due to the patients’ blood was collected at an early stage of disease development (before treatment), when the rate of multiple organ dysfunction in this group is still quite low (SOFA ≤ 2). Variations in SOFA scores between groups are significant due to the late manifestation of multiple organ dysfunction in COVID-19 patients, the opposite of the explosive course of the disease, which is a characteristic of bacterial sepsis9. But current hypotheses suggest a connection between CS, caused by COVID-19, and the occurrence of viral sepsis17. That’s why the comparison of patients with COVID-19 associated with CS and patients with SS seems reasonable.
Comorbidities can make the course of COVID-19 more severe and enhance CS18,19. In this regard, it would be interesting to determine how comorbidity would affect the results of comparing the metabolomic profiles of patients with COVID-19 and surgical septic patients.
This study is one of the first metabolomic studies using a biobank and a significant amount of samples from COVID-19 patients with cytokine storm and surgical sepsis from St. Petersburg and the Leningrad region (Russian Federation). The study was carried out on serum material collected strictly before the beginning of treatment, which allowed to obtain fairly “clean” serum samples without additional drug interference in the patients’ metabolome. The aim of this study was to compare the serum metabolomic profiles of patients with COVID-19-associated CS with the serum metabolomic profiles of septic patients after surgery.
Material and methods
Participants
Frozen blood serum from the collection of the biobank of St. Petersburg State Healthcare Establishment "City Hospital No. 40"was used. The study was conducted within the framework of the research project "Biobanking and biomedical research of human tissue and fluid samples" and was approved by the Expert Council on Ethics of St. Petersburg State Healthcare Establishment "City Hospital No. 40" (session No. 119, February 9, 2017). A total of 234 patients who underwent treatment at City Hospital No. 40 took part in the current retrospective study of the metabolomics profile. Samples were collected during hospital stays from January 2018 to March 2021 for septic patients and from May 2020 to June 2021 for COVID-19 patients. All patients underwent standard examinations according to clinical recommendations and diagnosis. Written informed consent was obtained from all patients for sample collection and storage in a biobank for subsequent use for scientific purposes and for the results’ publication. The study was conducted in accordance with the World Medical Association’s Code of Ethics (Declaration of Helsinki) for experiments involving humans. The patients were grouped as follows:
1.COVID(-): COVID-19 patients with CS without comorbidity (n = 40).
2.COVID( +): COVID-19 patients with CS with comorbidity (n = 43).
3. Sepsis: SS patients (n = 41).
4.Control: healthy volunteers (n = 110).
COVID-19 was diagnosed by a polymerase chain reaction (PCR) of nasopharyngeal swabs. CS was determined according to the following conditions: ferritin > 485 μg/L, C-reactive protein > 50 mg/L, d-dimer > 2.5 μg/mL, interleukin-6 > 25 pg/mL, LDH > 550 U/L.
Comorbidities were determined based on the patient’s self-report on admission and confirmed, if necessary, by further examinations. The Charlson Comorbidity Index (CCI) was calculated for each patient with COVID-1920. Patients with CCI ≤ 2 were included in the COVID(-) group, and patients with CCI ≥ 5 were included in the COVID( +) group.
The diagnosis of sepsis was made according to the Sepsis-3 consensus criteria and the SOFA scale5. The cause of sepsis in this group was bacterial infection as a complication after abdominal surgery.The Control group was selected during regular preventive check-ups. The criteria for inclusion in the group were the absence of COVID-19, confirmed with PCR, and the absence of sepsis. The general inclusion criterion was age over 18 years. Since our study is conducted in a real-world setting, we didn’t employ any additional inclusion or exclusion criteria.
Study design
The objective of this study was to compare the serum metabolomic profiles of patients with CS associated with COVID-19 and patients with sepsis. Metabolomic profiles were obtained using target LC–MS/MS analyses. The study solved the following tasks:
1) t-SNE clustering of patients.
2)Comparison of serum metabolomes of patients of the COVID(-) and COVID( +) groups: identification of common and differentially represented metabolites that alter levels relative to the Control group.
3)Identification of metabolites differentially represented in the serum of the Sepsis group compared to the Control.
4)Comparison of serum metabolomic profiles of patients of the Sepsis groups with serum metabolomes of patients in the COVID(-) and COVID( +) groups: identification of common and differentially represented metabolites.
Sample collection and storage
Blood samples from patients diagnosed with COVID-19 were collected within day of hospital admission and prior to treatment initiation. Blood samples from septic patients were collected when they were admitted to the intensive care unit (ICU) before starting antibiotic treatment. The blood samples for the Control group were obtained during a routine examination of volunteers. All samples were collected into Vacutest tubes (Gel and clot act., Vacutest Kima S.R.L., Italy). After centrifugation for 10 min at 4⁰C, 2200 rpm, the serum was collected and immediately frozen at -80⁰C. All samples were annotated, indicating the stage of the disease, gender, age, etc. Before analysis, the frozen samples were slowly warmed to room temperature and thoroughly mixed.
Metabolomic profiling of serum samples
The preparation of all blood serum samples for targeted metabolome studies was carried out in duplicate. 2-(N-morpholino)ethanesulfonic acid hydrate (MES) (CAS: 4432–31-9; cat no. M8250, Sigma-Aldrich) and L-methionine sulfone (CAS: 7314–32-1, cat No. M0876, Sigma-Aldrich) were used as internal standards. The samples were thawed at room temperature, and 100 μl of an ice-cold mixture of internal standards of a fixed concentration (25 μg/ml) in acetonitrile (cat no. 9012.2500GL, LC–MS-grade, J.T. Baker) was added to 50 μl of serum. The mixture was vortexes and incubated for 10 min at room temperature, then centrifuged (Centrifuge 5810R, Eppendorf) at 12,000 g for 10 min at 4 °C. An aliquot of 80 uL supernatant was diluted with Milli-Q—water acidified with formic acid (cat No. 533002, for LC–MS LiChropur, > 99%, Merck) to pH 2 and analyzed using the LC–MS/MS method. The prepared standard solutions and extracts were stored at -20 ⁰C.
Targeted metabolite profiling was performed using a liquid chromatography-mass spectrometer with a triple quadrupole LCMS-8050 (Shimadzu) along with a Nexera X2 (Shimadzu) chromatography system. The analysis was carried out following the established "LC/MS/MS Method Package for Primary Metabolites" by Shimadzu, utilizing the multiple reaction monitoring mode. This method allows simultaneous analysis of 98 analytes of the main chemical classes of clinically significant low-molecular compounds, including amino acids, organic acids, nucleotides, nucleosides, and coenzymes (Supplementary Table S1, S3). An analytical column Discovery HS F5-3 (150 × 2.1 mm, 3 mkm) (Supelco, Merck) and SecurityGuard “SupelGuard Discovery HS F5-3” (20 × 2.1 mm, 3 mkm, Supelco) were used to separate the analytes. Mass-spectrometry parameters and chromatographic conditions were meticulously set in accordance with the guidelines provided in the manual for the "LC/MS/MS Method Package for Primary Metabolites" method. Briefly, both ESI ( +) and ESI ( −) modes used water (LC–MS grade) as mobile phase (A) and ACN as mobile phase (B), and formic acid was used as mobile phase modifier. The gradient program was changed from 0 to 95% (B). Chromatographic and mass spectrometric parameters, as set out in the manual, are given in Supplementary Table S2. Data collection and processing were performed using LabSolutions software.
Metabolites were identified based on chromatographic retention time, m/z values of product ions, and their intensity ratios. Only chromatographic peaks with a peak-to-noise cutoff ratio ≥ 10 were considered. The content of the investigated substances was determined using the internal standard method, which considers the response (area) of the analyte connections in relation to the response (area) of the internal standard. All level measurements are given in arbitrary units of the content of the internal standard (arbitrary units — a.u.). The results correspond to the average of two parallel measurements of the same sample.
To ensure quality control (QA/QC), control samples were analyzed along with experimental samples to monitor instrument performance and facilitate chromatographic alignment. Control samples consisted of extracts of averaged blood serum samples with internal standards, prepared following the same procedure as the experimental samples. The averaged serum, derived from thoroughly mixed blood serum samples from seven donors, was aliquoted in 200 μl portions and frozen at -80 °C for later analysis. An appropriate volume of Milli-Q water passed through all stages of sample preparation was used as a “blank” sample. Additionally, all solvents used in sample preparation were also analyzed.
We utilized quality control samples to assess the reproducibility and stability of the prepared extracts on both intra- and inter-day bases. The internal standard solutions and control extracts were examined over a 10-day period, when they were stored at -20 °C between analyses and once at + 5 °C for 24 h. Overall process variability was determined by calculating the median RSD for IS and all endogenous metabolites (i.e., non-instrumental standards) present in control samples. The discrepancy between parallel measurements remained within 15% of the average values, with average daily deviations below 20%. Quality control samples were examined after every 50–60 experimental sample injections in order to ensure the consistency of the chromatographic retention times and responses of the studied compounds. Instrument variability was determined by calculating the median relative standard deviation (RSD) of the internal standards added to each sample during extraction. The scatter of the obtained values was evaluated during averaging, ensuring a maximum difference of 20% between the averaged parallel measurements. If this threshold was exceeded 25%, the sample was reanalyzed, and the initial result was disregarded.
Statistical analysis
To test the hypothesis of normal data distribution, the Shapiro–Wilk test was used. Data were transformed using Median & Quantile Absolute Deviation based Z-Score. The Z-Score transformation was applied solely to provide a more visually interpretable representation of the data in tabular form. To identify intergroup differences in the concentration levels of the studied metabolites, assessed through a.u., a nonparametric one-way analysis of variance was performed using the Kruskal–Wallis test; the Mann–Whitney test was used as a post-hoc analysis. The data was clustered using the t-distributed stochastic neighbor embedding (t-SNE) method, and the resulting data was visualized in two-dimensional space. The following parameters were used for the t-SNE analysis: Perplexity = 30, Learning Rate = 500, and Number of Iterations = 2000.
The log fold change (lfc) was used as a measure reflecting the difference in the range of values between samples; descriptive statistics are presented by the median (Me) and interquartile range [Q1-Q3]. The difference between samples was considered significant at p < 0.05 and |lfc|> 0.5. A Volcano plot was used to visualize the test results. Data processing and statistical analysis were performed using the R programming language version 4.3.1 and the Python programming language version 3.12. Spearman’s rank correlation coefficient was used for the correlation analysis. Metabolic pathway analysis was performed using the MetaboAnalyst 6.0 package and KEGG database21,22,23.
Results
Patient characteristics
Table 1 presents the demographic and clinical characteristics of the patient groups included in the study. The average age of comorbid patients was slightly higher than the average age in other groups (mean 74 ± 9 y vs. 51 ± 9.8 y for COVID(-), 65.2 ± 16 y for Sepsis and 48.1 ± 14 y for Control). The imbalance can be explained by the fact that comorbidities usually arise at a later age. The spectrum of diagnosis in COVID( +) group includes the illnesses related to the cardiovascular system, digestive system and liver function, renal function, neurology, diabetes mellitus and oncology.
The general characteristics of the changes in the serum metabolome of COVID-19 and sepsis groups
The levels of 100 compounds were studied using the LC/MS/MS procedure (Supplementary Table S1, S3). 83 compounds whose results were differed from zero were taken for the further analysis (Supplementary Table S4). The graphical result is presented on a heatmap (Fig. 1).

Heatmap of metabolites concentrations for each patient. Data normalized using median and quantile absolute deviation based Z-Score. Patients devided into groups: Sepsis, COVID(-), COVID( +) and Control. The borders between groups are designated by white lines across the heatmap.
There was a significant difference in age of the patients in the studied groups. To answer the question about the influence of age factor on the metabolites levels we conducted the correlation analysis (Supplementary Table S5). As all can see for most of the metabolites the correlation with age of the patients is closer to weak. The metabolites with most growing levels with age are cytosine, L-acetylcarnitine, DL-Dopa (ρ 0.45–0.48). The week decline of the level with age (ρ = -0.43) was shown for glutathione. t-SNE clustering of patients was performed based on the obtained metabolomic data (Supplementary Table S4). It revealed 3 clear clusters groups: Sepsis, Control and COVID-19 (Fig. 2). Interestingly, the COVID(-) and COVID( +) groups formed approximately one cluster, practically not separated. The Control group was notably segregated, situated far away from the other clusters. Additionally, data from sepsis patients was also well separated from others, while being in proximity to a cluster of COVID-19 patients.

t-SNE clustering of patients based on the LC–MS/MS analysis.
We conducted the correlation analysis of to investigate the point of changing the relations of metabolites in groups and found that COVID(-), COVID( +) and Sepsis groups differed significantly in functioning of metabolome which reflected in changing of correlation links between the groups (Fig. 3). It was rather unexpectedly because the t-SNE clustering demonstrated that COVID(-) and COVID( +) groups belonged to one cluster. t-SNE is a dimensionality reduction technique used to visualize high-dimensional data in two- or three-dimensional space. In this study, t-SNE was applied to group patients based on their metabolomic profiles. The clustering revealed clear separation among the Sepsis, Control, and COVID-19 groups. However, the COVID(-) and COVID( +) groups formed a single cluster, indicating global similarity in their metabolomic profiles.

Correlation analysis of the metabolites concentrations in groups. The color of each cell represents the level of Spearman’s rank correlation coefficient from -1 to 1. A. COVID(-) group. B. COVID( +) group. C. Sepsis group.
Correlation analysis, on the other hand, examined the relationships between individual metabolites, showing how they covary within each group. This analysis revealed significant differences in the correlation patterns of metabolites between the COVID(-), COVID( +), and Sepsis groups, highlighting distinct metabolic interactions in each group. These findings were unexpected, given that t-SNE clustering placed the COVID(-) and COVID( +) groups within the same cluster. These results are not contradictory, as each method evaluates different aspects of the data. t-SNE clustering focuses on global similarities in metabolomic profiles, while correlation analysis highlights subtle differences in the functional organization and metabolic interactions. Thus, despite the overall similarity in COVID(-) and COVID( +) profiles as indicated by t-SNE, correlation analysis reveals that the metabolic relationships within these groups are likely influenced by comorbidities.
COVID(-) vs. COVID( +) groups metabolomics
We compared the metabolomic profiles of COVID(-) and COVID( +) patients (Supplementary Table S4, Fig. 4, 5 ). 62 and 67 metabolites were changed compared to the Control in COVID(-) and COVID( +) groups (p < 0.05), respectively. A decline of 38 metabolites along with rise of 24 compounds was recorded in COVID(-) serum; the most prominent changes are presented in Fig. 4AD. While for COVID( +) group, 45 metabolites showed a decrease and 22 metabolites exhibited an increase compared to the Control group (Fig. 4 BD ). Among 9 compounds mostly increased in the COVID(-) group, 6 metabolites were also risen in the COVID( +) group. These were dimethylglycine, L-acetylcarnitine, L-kynurenine, L-phenylalanine, L-cystathionine, adenosine monophosphate (Fig. 4 AB). Among 25 metabolites mostly decreased in the COVID( +) group, the levels of 17 compounds also fell in the COVID(-) group. These were L-histidine, citrulline, ornithine, uridine, uric acid, L-arginine, asymmetric dimethylarginine, pantothenic acid, L-threonine, 4-hydroxyproline, choline, allantoin, inosine, glycine, L-leucine, acetylcholine chloride, L-isoleucine (Fig. 4 AB).

Main abnormal metabolites in COVID(-), COVID( +) and Sepsis group in comparison with the Control. Volcano plots parametres: Log2 fold change cutoff 0.5; p-value cutoff 0.05. A. COVID(-) vs Control. B. COVID( +) vs Control. C. Sepsis vs Control. D. Heat map of concentration of metabolites in COVID(-), COVID( +) and Sepsis groups. The color of each cell represents logarithm of fold change of metabolites with concentration of metabolites in control group, and p values were calculated using Mann–Whitney U test, *p < 0.05, **p < 0.01, and ***p < 0.001. E. The Venn diagram shows the overall quantity of differentialy abundant in comparison with Control metabolites in COVID(-), COVID( +) and Sepsis group highlighting 47 metabolites common to all, 7 metabolites common to COVID(-) and COVID( +), 6 common to COVID(-) and Sepsis, 10 common for COVID( +) and Sepsis, 1 unique for COVID( +) and 11 unique for Sepsis.

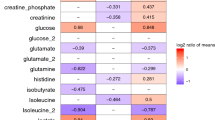
Comparison of the COVID(-), COVID( +) and Sepsis groups. The volcano plots show most significant differentially abundant metabolites. Log2 fold change cutoff 0.5; p-value cutoff 0.05. A. COVID(-) vs COVID( +). B. Sepsis vs COVID(-). C. Sepsis vs COVID( +).
In spite of the presence of some common characteristics of metabolomics changes, there were 29 compounds, which revealed significant (p < 0.05) differences in the levels between the COVID(-) and COVID( +) groups (Supplementary Table S4, Fig. 5A ). 15 metabolites exhibited higher levels in the COVID( +) group compared to the COVID(-) group. L-kynurenine, L-lactic acid, L-alanine, uric acid, uracil, carnosine, ornithine, and norepinephrine were the most prominent. Conversely, 14 metabolites showed decreased levels in the COVID( +) group, with L-proline and serine being the most notable (Fig. 5A). Interestingly, some of these compounds changed their levels in series from Control to COVID(-) and then to COVID( +) group. For example, L-kynurenine and ornithine revealed such dynamics (Supplementary Table S4).
To evaluate the potential metabolomic shifts that resulted from changes in measured compounds, an enriched pathway analysis was held. We identified the top 8 common pathways from the KEGG database that were most significantly dysregulated in the COVID(-) and COVID( +) groups compared with the Control group (Fig. 6 AB; Supplementary Table S6 AB). The four of these pathways were uniquely disturbed in each group (Fig. 3 C; Supplementary Table S5 D). The metabolism of cysteine and methionine showed the largest difference, being more disrupted in COVID( +) vs. COVID(-) patients (Fig. 7A; Supplementary Table S6 AB).

The main KEGG pathways disturbed in groups. A – C: the pathways view according to the p-values and the pathway impact values. Dots present the most relevant pathways. The pathway impact is presented by size and p-value by color. A. COVID(-) vs. Control. B. COVID( +) vs. Control. C. Sepsis vs. Control. D – F: a summary plots for quantitative enrichment analysis showing the top 25 enriched terms for each group. D. COVID(-). E. COVID( +). F. Sepsis. KEGG pathways were identified and visualized using the KEGG database (Kanehisa Laboratories, www.kegg.jp/kegg/kegg1.html). Permission to use KEGG pathways was obtained from Kanehisa Laboratories.

The main KEGG pathways disturbed between groups. A – C: the pathways view according to the p-values and the pathway impact values. Dots present the most relevant pathways. The pathway impact is presented by size and p-value by color. A. COVID(-) vs. COVID( +). B. COVID(-) vs. Sepsis. C. COVID( +) vs. Sepsis. D – F: a summary plots for quantitative enrichment analysis showing the top 25 enriched terms for each group. D. COVID(-) vs. COVID( +). E. COVID(-) vs. Sepsis. F. COVID( +) vs. Sepsis. KEGG pathways were identified and visualized using the KEGG database (Kanehisa Laboratories, www.kegg.jp/kegg/kegg1.html). Permission to use KEGG pathways was obtained from Kanehisa Laboratories.
These data, in consistency with t-SNE clustering (p. 3.2), demonstrated that the difference between COVID( +) and COVID(-) groups was not substantial and had rather quantitative than qualitative character.
Sepsis group metabolomics
In comparing the Sepsis and Control groups, we found differences in the levels of 75 metabolites (p < 0.05) (Supplementary Table S4, Fig. 4 CD). Among these metabolites, 35 exhibited elevated levels and 40 metabolites displayed decreased levels. Enriched pathway analysis identified the top 10 pathways in the KEGG database that were potentially disturbed in the Sepsis group (Fig. 6 C; Supplementary Table S6 C). These pathways included arginine biosynthesis; cysteine and methionine metabolism; alanine, aspartate, and glutamate metabolism; glycine, serine, and threonine metabolism; citrate (TCA) cycle, arginine and proline metabolism; pyrimidine metabolism; tyrosine metabolism; histidine metabolism, and glutathione metabolism.
Sepsis vs. COVID-19 groups metabolomics
Comparison of the Control group with the Sepsis, COVID(-), and COVID( +) groups indicated significant differences in metabolite abundance (p ≤ 0.05). Among the groups, the greatest number of changes in metabolite levels (74) was observed in the Sepsis vs. Control comparison. In the comparison of COVID(-) vs. Control, 60 metabolites showed varying levels, while 65 metabolites displayed differences in the COVID( +) vs. Control comparison (Supplementary Table S4, Fig. 4 E).
The levels of 53 metabolites changed significantly (p ≤ 0.05) both for COVID(-) and Sepsis groups relative to the Control group. Within these groups, the levels of 11 metabolites increased, and 22 metabolites decreased in both groups. Similarly, the levels of 57 metabolites showed significant changes in both the COVID( +) and Sepsis groups relative to the Control group; the levels of 14 metabolites increased and 25 metabolites decreased in both groups (Supplementary Table S4). The levels of 47 metabolites changed in all three groups. All three groups exhibited increased levels of 11 metabolites relative to the Control. The most prominent were dimethylglycine, L-acetylcarnitine, L-cystathionine, adenosine monophosphate. Additionally, 19 metabolites displayed lower levels compared to the controls across all groups. The most prominent are L-histidine, citrulline, ornithine, uridine, pantothenic acid, L-threonine, choline, inosine, L-leucine, and L-isoleucine (Fig. 4 ABC ).
Comparison of the metabolomic profiles of the Sepsis and COVID(-) groups showed that the levels of 60 metabolites were significantly different (p < 0.05) (Supplementary Table S4). Among these, the levels of 38 metabolites showed higher levels in the Sepsis group, and 22 metabolites displayed lower levels in the Sepsis group. The most significant changes were displayed on Fig. 5B.
The comparison of metabolomic profiles between the Sepsis and COVID( +) groups showed that the levels of 56 metabolites were significantly different (p < 0.05) (Supplementary Table S4). Of these, in the Sepsis group, the levels of 36 metabolites were higher and the levels of 20 metabolites were lower. The most significant changes were displayed on Fig. 5C.
The levels of 36 metabolites were significantly different (p ≤ 0.01) in both COVID-19 groups relative to the Sepsis group; of these, the levels of 28 metabolites were higher in the serum of septic patients (Supplementary Table S4, Fig. 5 BC). The most prominent were acetylcholine chloride, L-histidine, uric acid, allantoin, 4-hydroxyproline, asymmetric dimethylarginine, creatine, creatinine, citric acid, methionine sulfoxide, guanosine, L-carnitine, L-cystathionine, carnosine. The levels of 12 compounds were lower in the serum of septic patients compared to COVID-19 one. The most prominent were L-proline, L-aspartic acid, L-tryptophan, niacinamide, L-tyrosine, L-phenylalanine, L-glutamic acid (Fig. 5 BC).
Some metabolites levels changed solely in the Sepsis or COVID-19 groups. So as 4-hydroxyproline, isocitric and pyruvic acids, procollagen-5-hydroxylisine, creatine, creatnine, SAM, acetylcholine chlorid, cytric acid, serotonine, symmetric dimethiylarginine significantly increased, and L-aspartic acid, L-glutamic acid, L-proline, L-lysine, GABA, niacinamide significantly decreased exclusively in the Sepsis group. In contrast to the Sepsis group, both groups of patients with COVID-19 were characterized by a significant increase in citicoline, 5-thymidilic acid, GABA, nicotinic acid, L-phenylalanine, histamine and a decrease in allantoin, 4-hydroxiproline, isocytric and pyruvic acids, procollagen-5-hydroxylisine, acetylcholine chloride, symmetric and asymmetric dimethylarginine, L-methionine, uric acid, and L-lactic acid (Supplementary Table S4).
Enriched pathway analysis of the KEGG databases identified eight metabolic pathways, the differences in which for surgical septic and COVID-19 (COVID(-) and COVID( +) patients (p ≤ 0.05) (Fig. 7 BC ; Supplementary Table S6 EF). These pathways remained consistent when comparing Sepsis with both COVID(-) and COVID( +). There were cysteine and methionine metabolism, histidine metabolism, arginine and proline metabolism, arginine biosynthesis pathway, aspartate, glutamate, and alanine metabolism, phenylalanine, tyrosine, and tryptophan biosynthesis pathways, phenylalanine metabolism, and pyrimidine metabolism.
Discussion
We compared the serum metabolome of patients with COVID-19 related CS vs. surgical sepsis. Both syndromes stem from infections, with COVID-19 linked to a viral infection and surgical sepsis to a bacterial one. These conditions can lead to an increased inflammatory response that poses significant risks to patients. We carried out this study to contribute to the understanding of the pathophysiology underlying these hyperinflammatory processes.
The presence of comorbidities can make the course of COVID-19 more severe18,19. To investigate the impact of concomitant illnesses on metabolomic alterations two groups of CS patients were created: one without comorbidities (COVID(-)) and the other with comorbidities (COVID( +)). The metabolomic profiles of these groups were also compared.
Our research revealed parallel shifts in the metabolome of both the COVID( +) and COVID(-) groups. Levels of almost all amino acids, including proteogenic and glycogenic types, as well as key markers of energy metabolism such as pyruvate, lactate, and TCA cycle acids were significantly decreased. Other researchers have also reported decreased levels of these metabolites in patients with CS24,25. Furthermore, an association was found between the increase in plasma cytokines and the decrease in amino acid levels26. In this study, patients experienced more significant changes in certain energy-related metabolites within the COVID( +) group, where the IL6 level was elevated. Both COVID-19 groups were characterized by decreased carnitine along with increased acetylcarnitine levels, which might indicate an energy deficiency and impaired transport in the mitochondria. Similar changes have been reported by other authors27,28,29. Also a reduction in the levels of arginine, citrulline and ornithine indicates a significant disturbance in nitrogen metabolism in COVID-19 patients, aligning with findings from other researchers30,31. The pathway of tryptophan degradation linked to inflammation modulation underwent a common alteration in both groups of COVID-19, shifting towards kynurenine synthesis. These findings of increased kynurenine levels and decreased tryptophan and serotonin levels are consistent with existing data32. In COVID-19 patients, elevated levels of some other metabolites linked to the inflammatory response were observed. These included the inflammatory mediator histamine and the neurotransmitter gamma-aminobutyric acid (GABA), which is known to have anti-inflammatory properties32. We assumed that the increase in GABA levels was compensatory. The difference between the COVID(-) and COVID( +) groups can be considered insignificant. The most significant variations in the dynamics of changes were observed for L-proline and serine. Their levels increased sharply in the COVID(-) group and approached control levels in the COVID( +) group. It could be assumed that the changes in these amino acids in patients with comorbidities were related to the characteristics of the accompanying illnesses and their therapies. So we can propose that the main changes in the metabolomic profiles in both groups of COVID-19 patients were due to the virus infection, not to the comorbidities. This proposal is in line with the conclusion in33. Nevertheless, an interesting feature is related to the correlation analysis, which showed that the ratios of the studied metabolites were different in COVID(-) and COVID( +) groups. We suppose that it was the influence of comorbidity on the functioning of organism—with less changes levels we saw significant discrepancies in connections between metabolites in COVID(-) and COVID( +) groups.
The obtained data on the metabolomics of surgical septic patients, presented in Supplementary Table S4 and Fig. 4C, were consistent with the data of other researchers. This change in the levels of proteogenic and glycogenic amino acids was considered to be one of the characteristic features of sepsis34. The decrease in serum amino acids levels was explained by their use as substrate for the TCA cycle and glycolysis for energy demand, which greatly increased during sepsis11. The energy imbalance in the Sepsis group was confirmed in our study by increased levels of oxoglutarate and citrate, metabolites of the TCA cycle. These data are consistent in particular with studies presented in articles8,35.
Another feature of sepsis is a violation of beta-oxidation of fatty acids in mitochondria and an increase in acetylcarnitines levels12,36. In our study, L-acetylcarnitine was increased in surgical septic patients as in37. In addition, surgical septic patients had elevated kynurenine levels and decreased tryptophan and serotonin levels, suggesting a redirection of tryptophan degradation toward kynurenine synthesis. These results were consistent with data presented in12. The inflammatory marker procollagen 5-hydroxy-L-lysine was also significantly increased in the Sepsis group. Similar findings were reported in38. The increase in the levels of dopamine as in39 and acetylcholine chloride that we found could be attributed to the compensatory anti-inflammatory effect of these metabolites40,41. In addition, we detected a significant change in the levels of compounds associated with the development of oxidative stress in surgical septic patients—decreased levels of glutathione and increased levels of its precursor gamma-glutamylcysteine, ophthalmic acid and symmetric dimethylarginine, as noted by other authors11,42,43. We should note that there is not enough information in the literature on the levels of oxoglutaric acid and acetylcholine chloride in septic serum or plasma. Therefore, our results for these metabolites were among the first and needed confirmation. Overall, the metabolomic profile data of patients with COVID-19 and surgical sepsis were in agreement with findings from other researchers.
We observed a similarity in the direction of level changes for various metabolites when comparing the metabolomic profiles of patients with COVID-19 and surgical sepsis, across the Control—COVID(-)—COVID( +)—Sepsis series (Fig. 4 ABCD ). For example, the levels of L-alanine, ornithine and uric acid increased sequentially. It could be linked to a higher breakdown of proteins and nucleic acids caused by cell death and inflammation due to a more severe course of the pathological process in the COVID( +) group than in the COVID(-) group and more inflammation and oxidative stress in the Sepsis group than in COVID-19 patients11,44. An increase in uric acid levels was also a marker of gradual deterioration of renal function in the Control—COVID(-)—COVID( +)—Sepsis series45. This was confirmed by clinical analysis of patients’ creatinine levels (Table 1).
The inflammatory marker kynurenine showed elevated levels in all three groups. Activation of the kynurenine pathway with increased levels of kynurenine and other participants in this pathway is associated with immunosuppression in response to inflammatory signaling46. In the Sepsis group its level was lower than in the COVID(-) group, but the difference is insignificant, so making it difficult to draw any conclusions on this point. While in the COVID( +) group the kynurenine level was the highest, presumably with more severe inflammation. It is known that the kynurenine pathway of tryptophan degradation is activated by high levels of proinflammatory cytokines, in particular IFNγ and IL647. Based on the data in Table 1, we can assume that the higher level of IL6 in the COVID( +) group indicates a greater degree of activation of the kynurenine pathway. Some comorbidities, such as the most common cardiovascular problems, are associated with inflammation and activation of the kynurenin pathway45. Therefore, we can assume that the comorbidities in the COVID( +) group contributed to the elevated kynurenin levels. Another participant in the tryptophan degradation pathway, serotonin, showed the opposite alteration. Its level was highest in the Sepsis group and lowest in the COVID( +) group. This fact requires further investigation to better understanding of the involvement of the kynurenine pathway in hyperinflammatory conditions of various origins.
The level of the antioxidant carnosine increased sequentially in the COVID(-)—COVID( +)—Sepsis series. Moreover, its level was lower in the COVID(-) group than in the Control group, but significantly higher in the COVID( +) and Sepsis groups. Also in the COVID(-), COVID( +) and Sepsis series, there was a consistent increase in the levels of norepinephrine, a metabolite known for its reported anti-inflammatory properties48. At the same time, the levels of the norepinephrine precursor in the synthesis pathway, L-tyrosine, changed in the opposite direction and decreased proportionally across the series. This suggests that there may be compensatory production of carnosine and norepinephrine as inflammation and oxidative stress escalated within the sequence Control—COVID(-)—COVID( +)—Sepsis.
The sequential increase in lactic acid in the COVID(-)—COVID( +)—Sepsis series appeared to be explicable. As lactic acid levels increased in the groups studied, it indicated a transition towards glycolysis, signaling a shift in energy metabolism. In all three groups, it was noteworthy that the level of lactic acid remained lower compared to the Control group, despite documented elevations in lactic acid levels observed in sepsis and severe cases of COVID-1949,50.
Significant changes in methionine metabolism were observed in all groups. In the progression from COVID(-) to COVID( +) to Sepsis, a notable change in the concentrations of two metabolites associated with the methionine cycle was detected: an increase in SAH levels and a decrease in serine levels. It should be noted that a decrease in serine levels could affect both methionine synthesis and the pathway for the production of the antioxidant glutathione, which showed similarly low levels in all three groups. In the progression from COVID(-) to COVID( +) to Sepsis, methionine sulfoxide, another participant in the methionine cycle, showed a steady increase in levels. We assumed that this was necessary to maintain the level of methionine as a key metabolite in many processes.
Levels of several metabolites fell to a similar extent in patients with COVID-19 and in the Sepsis group (Fig. 4 ABCD). Firstly, it is the decrease in amino acid levels observed in both the Sepsis group and patients with COVID-19. This decrease in amino acid levels may indicate an increased demand for oxidative sources to energy cycles in all groups24. Levels of glutathione and choline, known for their ability to decrease oxidative stress and inflammation, diminished equally in individuals with surgical sepsis and with COVID-19.
So, while there were resemblances in the metabolomic profiles, the distinctions in them for Sepsis and COVID-19 patients were also significant. Figure 4E schematically demonstrated the number of metabolites whose levels changed significantly in the three groups. The greatest number of such metabolites belonged to the Sepsis group (74); the COVID(-) and COVID( +) groups differed slightly in this feature (60 and 65 metabolites, respectively), but the difference between both COVID-19 groups and Sepsis group was much more.This could be explained by the different dynamics of both pathological processes. As already mentioned, sepsis is characterized by a more explosive course than CSS, which also leads to severe consequences, but more slowly43.
For the intergroup comparison of Sepsis vs. COVID-19, 28 metabolites showed a significant increase (p ≤ 0.01) compared to both groups of COVID-19 patients (p. 3.4). These included metabolites related to mitochondrial function, markers of oxidative stress, collagen destruction, methionine and transsulfuration cycles, and renal failure. Most striking were the differences in the levels of metabolites in the TCA cycle. This probably reflects the biological basis of both syndromes. COVID-19 is characterized in most cases with lung injury at the beginning of the infection and oxygen deficiency as a consequence. Thus, suppression of the TCA cycle occurred. Surgical sepsis is not necessarily associated with hard lung injury44, especially in the early stage, as in our patients. In the absence of hypooxigenation, the TCA cycle is upregulated to meet the increased demand for energy production to maintain hyperinflammation and immune function.
Some metabolites exhibited a significant (p ≤ 0.01) downregulation in surgical septic patients compared to COVID-19 patients (Fig. 5 BC). The lower levels of several amino acids in the Sepsis group could be associated with the greatest need for energy and more intensive utilization of amino acids in energy cycles. However, the most significant changes were observed in the profiles of GABA and niacinamide. These metabolites were increased in the COVID(-) and COVID( +) groups, but their levels decreased by several orders of magnitude in the Sepsis group. GABA may have an anti-inflammatory function30, and niacinamide was the precursor in the synthesis of NAD. Such alterations might indicate a significantly higher level of inflammation and energy problems in surgical septic patients compared to patients with COVID-19.
To evaluate the potential changes in metabolomic pathways, we performed an enriched analysis of the KEGG databases. The analysis revealed that certain pathways common to the COVID(-), COVID( +), and Sepsis groups were significantly perturbed. These pathways included: glycine, serine and threonine metabolism; arginine metabolism; cysteine and methionine metabolism; arginine and proline metabolism; alanine, aspartate and glutamate metabolism; glutathione metabolism; tyrosine metabolism; and histidine metabolism (Fig. 6). Among these, the cysteine and methionine pathway was one of the most altered in all groups. By evaluating the degree of alteration in this pathway based on the number of metabolites analyzed, the p-value, and the impact factor, one could see a greater impairment in the COVID( +) and Sepsis groups than in the COVID(-) group. The cysteine and methionine pathway is one of the most important in the body. It is associated both with maintaining the level of methionine, which is a donor of methyl groups, and with the transformations of sulfur-containing amino acids responsible for redox potential. In our study, the pathways of arginine biosynthesis and histidine metabolism were also similarly altered in all three groups. The TCA cycle was disturbed only in the COVID( +) and Sepsis groups, but to a greater extent in the Sepsis group, as assessed by the number of metabolites analyzed, p-value and impact factor. This analysis confirmed that significant metabolic changes in the serum of COVID-19 and surgical septic patients were primarily linked to amino acid metabolism and alterations in redox potential and energy cycle, with metabolic irregularities amplifying from COVID(-) to COVID( +) to Sepsis.
As we mentioned in Sect. “Patient characteristics”, such important characteristics as SOFA score differed between the COVID-19 and Sepsis groups. This difference was due to the specificity of the collection of biological material from COVID-19 patients, as we tried to collect the material before the beginning of treatment. In this context the question arises: did this SOFA discrepancy influence the results? Several investigations of other scientists revealed that some metabolites, connected with inflammation and oxygen deficiency were growing in accordance with the severity of COVID-1928,30,51,52. On the basis of this knowledge we may carefully suppose that in unreal case of presence of the group of untreated patients with SOFA like in Sepsis group the results will be similar to those obtained in this work or more differentiating the groups in relation of metabolites of oxygen energetics or inflammation due in particular to more lowering the TCA metabolites and growth the levels of metabolites of kynurenin pathway in COVID-19 patients.
This study had several limitations. First, we performed a targeted metabolomic study, so our results did not cover metabolomic changes as completely as a non-targeted metabolomic study can. Secondly, there was a substantial disparity in SOFA levels between the COVID-19 and Sepsis groups. This incongruity arose from our deliberate collection of blood samples from COVID-19 patients prior to treatment initiation to mitigate the impact of antibiotics, glucocorticoids, and similar factors. So, the observed SOFA discrepancy appeared due to the slower progression of multiple organ failure in COVID-19 compared to surgical sepsis. Finally, our study reflected real-world conditions, resulting in differences in age parameters between the compared groups. It is imperative to consider these limitations when evaluating the results of this research.
Conclusion
This study used biobank samples of COVID-19 patients with cytokine storm and surgical sepsis from St. Petersburg and the Leningrad region (Russian Federation) and represented one of the first comparative serum metabolomic studies in this geographical area. The serum of patients with COVID-19 and surgical sepsis showed significant changes in various metabolites linked to amino acid metabolism, nitrogen metabolism, inflammation, folate and methionine cycles, and glycolysis. The differences between the COVID(-) and COVID( +) groups were not significant. Changes in metabolite levels tended to increase consistently from COVID(-) to COVID( +) to Sepsis groups, with more pronounced changes in the Sepsis group. The most significant differences between surgical septic and COVID-19 patients appeared in metabolites related to kynurenine synthesis, niacinamide, the TCA cycle and GABA. In all groups, there were significant alterations in cysteine and methionine metabolic pathways. So, our study revealed common and different features of the metabolomic profiles of patients with surgical sepsis and CS associated with COVID-19.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
WHO. WHO Coronavirus (COVID-19) Dashboard WHO: 2024. https://data.who.int/dashboards/ COVID-19/deaths, 2024 (accessed 14 January 2024)
Fajgenbaum, D. C. & June, C. H. Cytokine storm. N. Engl. J. Med. 383, 2255–2273. https://doi.org/10.1056/NEJMra2026131 (2020).
Cron, R. Q., Goyal, G. & Chatham, W. W. Cytokine storm syndrome. Annu. Rev. Med. 74, 321–337. https://doi.org/10.1146/annurev-med-042921-112837 (2023).
Jarczak, D. & Nierhaus, A. Cytokine storm-definition, causes, and implications. Int. J. Mol. Sci. 23, 11740. https://doi.org/10.3390/ijms231911740 (2022).
Singer, M. et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 315, 801–810. https://doi.org/10.1001/jama.2016.0287 (2016).
Dyck, B., Unterberg, M., Adamzik, M. & Koos, B. The impact of pathogens on sepsis prevalence and outcome. Pathogens. 13, 89. https://doi.org/10.3390/pathogens13010089 (2024).
Sepsis Alliance. Sepsis and TOPIC. https://www.sepsis.org/sepsisand/coronavirus-covid-19/. 2023 (accessed 04 March 2024).
Lin, S. H. et al. Exploring plasma metabolomic changes in sepsis: a clinical matching study based on gas chromatography-mass spectrometry. Ann. Transl. Med. 8, 1568. https://doi.org/10.21037/atm-20-3562 (2020).
Kellum, J. A., Nadim, M. K. & Forni, L. G. Sepsis-associated acute kidney injury: is COVID-19 different?. Kidney Int. 98, 1370–1372. https://doi.org/10.1016/j.kint.2020.08.009 (2020).
Leisman, D. E. et al. Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 8, 1233–1244. https://doi.org/10.1016/S2213-2600(20)30404-5 (2020).
Su, L. et al. Dynamic changes in amino acid concentration profiles in patients with sepsis. PLoS One. 10, e0121933. https://doi.org/10.1371/journal.pone.0121933 (2015).
Beloborodova, N. V., Olenin, A. Y. & Pautova, A. K. Metabolomic findings in sepsis as a damage of host-microbial metabolism integration. J. Crit. Care. 43, 246–255. https://doi.org/10.1016/j.jcrc.2017.09.014 (2018).
Pandey, S. Sepsis, management and advances in metabolomics. Nanotheranostics. 8, 270–284. https://doi.org/10.7150/ntno.94071 (2024).
Bruzzone, C. et al. Metabolomics as a powerful tool for diagnostic, pronostic and drugintervention analysis in COVID-19. Front. Mol. Biosci. 10, 1111482. https://doi.org/10.3389/fmolb.2023.1111482 (2023).
Wang, T. et al. COVID-19 metabolism: Mechanisms and therapeutic targets. MedComm 3, e157. https://doi.org/10.1002/mco2.157 (2022).
Chen, P. et al. Metabolic alterations upon SARS-CoV-2 infection and potential therapeutic targets against coronavirus infection. Sig. Transduct. Target Ther. 8, 237. https://doi.org/10.1038/s41392-023-01510-8 (2023).
Li, H. et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 395, 1517–1520. https://doi.org/10.1016/S0140-6736(20)30920-X (2020).
Guan, W. J. et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: a nationwide analysis. Eur. Respir. J. 55, 2000547. https://doi.org/10.1183/13993003.00547-2020 (2020).
Chatterjee, S. et al. Association of COVID-19 with comorbidities: an update. ACS Pharmacol. Transl. Sci. 6, 334–354. https://doi.org/10.1021/acsptsci.2c00181 (2023).
Charlson, M. E., Pompei, P., Ales, K. L. & MacKenzie, C. R. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J. Chronic. Dis. 40, 373–383. https://doi.org/10.1016/0021-9681(87)90171-8 (1987).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Masoodi, M. et al. Disturbed lipid and amino acid metabolisms in COVID-19 patients. J. Mol. Med. (Berl). 100, 555–568. https://doi.org/10.1007/s00109-022-02177-4 (2022).
Gama-Almeida, M. C. et al. Integrated NMR and MS analysis of the plasma metabolome reveals major changes in one-carbon, lipid, and amino acid metabolism in severe and fatal cases of COVID-19. Metabolites. 13, 879. https://doi.org/10.3390/metabo13070879 (2023).
Ceballos, F. C. et al. Metabolic profiling at COVID-19 onset shows disease severity and sex-specific dysregulation. Front. Immunol. 13, 925558. https://doi.org/10.3389/fimmu.2022.925558 (2022).
Thomas, T. et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight. 5, e140327. https://doi.org/10.1172/jci.insight.140327 (2020).
Barberis, E. et al. Large-scale plasma analysis revealed new mechanisms and molecules associated with the host response to SARS-CoV-2. Int. J. Mol. Sci. 21, 8623. https://doi.org/10.3390/ijms21228623 (2020).
Costanzo, M. et al. COVIDomics: the proteomic and metabolomic signatures of COVID-19. Int. J. Mol. Sci. 23, 2414. https://doi.org/10.3390/ijms23052414 (2022).
Jia, H. et al. Metabolomic analyses reveal new stage-specific features of COVID-19. Eur. Respir. J. 59, 2100284. https://doi.org/10.1183/13993003.00284-2021 (2022).
Özbay, S. et al. Urea cycle and arginine metabolic changes in COVID-19 patients. Eurasian J. Emerg. Med. 22, 203–208. https://doi.org/10.4274/eajem.galenos.2023.75875 (2023).
Fu, J. et al. GABA regulates IL-1β production in macrophages. Cell Rep. 41, 111770. https://doi.org/10.1016/j.celrep.2022.111770 (2022).
Shi, D. et al. The serum metabolome of COVID-19 patients is distinctive and predictive. Metabolism. 118, 154739. https://doi.org/10.1016/j.metabol.2021.154739 (2021).
Oh, T. S. et al. Dichloroacetate improves systemic energy balance and feeding behavior during sepsis. JCI Insight. 7, e153944. https://doi.org/10.1172/jci.insight.153944 (2022).
Langley, R. J. et al. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci. Transl. Med. 5, 195. https://doi.org/10.1126/scitranslmed.3005893 (2013).
Amunugama, K., Pike, D. P. & Ford, D. A. The lipid biology of sepsis. J. Lipid Res. 62, 100090. https://doi.org/10.1016/j.jlr.2021.100090 (2021).
Schmerler, D. et al. Targeted metabolomics for discrimination of systemic inflammatory disorders in critically ill patients. J. lipid Res. 53, 1369–1375. https://doi.org/10.1194/jlr.P023309 (2012).
Gäddnäs, F. et al. Markers of collagen synthesis and degradation are increased in serum in severe sepsis: a longitudinal study of 44 patients. Crit. Care. 13, R53. https://doi.org/10.1186/cc7780 (2009).
Chen, et al. Integrative analysis of metabolomics and proteomics reveals amino acid metabolism disorder in sepsis. J. Transl. Med. 20, 123. https://doi.org/10.1186/s12967-022-03320-y (2022).
Jeremias, I. C. et al. The role of acetylcholine in the inflammatory response in animals surviving sepsis induced by cecal ligation and puncture. Mol. Neurobiol. 53, 6635–6643. https://doi.org/10.1007/s12035-015-9538-y (2016).
Moore, S. C., Vaz de Castro, P. A. S., Yaqub, D., Jose, P. A. & Armando, I. Anti-inflammatory effects of peripheral dopamine. Int. J. Mol. Sci. 24, 13816. https://doi.org/10.3390/ijms241813816 (2023).
Soga, T. et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 281, 16768–16776. https://doi.org/10.1074/jbc.M601876200 (2006).
Winkler, M. S. et al. Symmetrical (SDMA) and asymmetrical dimethylarginine (ADMA) in sepsis: high plasma levels as combined risk markers for sepsis survival. Crit. Care. 22, 216. https://doi.org/10.1186/s13054-018-2090-1 (2018).
Koçak, T. Z., Kayaaslan, B. & Mer, M. COVID-19 and sepsis. Turk. J. Med. Sci. 51, 3301–3311. https://doi.org/10.3906/sag-2108-239 (2021).
Johnson, R. J., Sanchez Lozada, L. G., Lanaspa, M. A., Piani, F. & Borghi, C. Uric acid and chronic kidney disease: still more to do. Kidney Int. Rep. 8, 229–239. https://doi.org/10.1016/j.ekir.2022.11.016 (2022).
Dehhaghi, M. et al. The roles of the kynurenine pathway in COVID-19 neuropathogenesis. Infection. 52, 2043–2059. https://doi.org/10.1007/s15010-024-02293-y (2024).
Yang, Y. et al. The role of the kynurenine pathway in cardiovascular disease. Front. Cardiovasc. Med. 11, 1406856. https://doi.org/10.3389/fcvm.2024.1406856 (2024).
Thoppil, J., Mehta, P., Bartels, B., Sharma, D. & Farrar, J. D. Impact of norepinephrine on immunity and oxidative metabolism in sepsis. Front. Immunol. 14, 1271098. https://doi.org/10.3389/fimmu.2023.1271098 (2023).
Icard, P. et al. The key role of Warburg effect in SARS-CoV-2 replication and associated inflammatory response. Biochimie. 180, 169–177. https://doi.org/10.1016/j.biochi.2020.11.010 (2021).
Liu, J., Zhou, G., Wang, X. & Liu, D. Metabolic reprogramming consequences of sepsis: adaptations and contradictions. Cell. Mol. Life Sci. 79, 456. https://doi.org/10.1007/s00018-022-04490-0 (2022).
Valdés, A. et al. Metabolomics study of COVID-19 patients in four different clinical stages. Sci. Rep. 12, 1650. https://doi.org/10.1038/s41598-022-05667-0 (2022).
Gu, M. et al. Sera metabolomics characterization of patients at different stages in Wuhan identifies critical biomarkers of COVID-19. Front. Cell. Infect. Microbiol. 12, 882661. https://doi.org/10.3389/fcimb.2022.882661 (2022).
Funding
Supported by Saint Petersburg State University, project ID: 124032900005-4.
Author information
Authors and Affiliations
Contributions
Russkikh Ia.V.: Investigation, validation, data curation, writing, editing. Popov O.S.: Conceptualization, investigation, data curation, formal analysis, visualization, writing, editing. Klochkova T.G.: conceptualization, writing, editing, corresponding author. Sushentseva N.N.: Conceptualization, methodology, writing, editing. Apalko S.V.: Conceptualization, project administration, writing, editing. Asinovskaya A.Yu.: Resources, project administration, supervision. Mosenko S.V.: Data curation, resources. Sarana A.M.: Supervision, project administration. Shcherbak S.G.: Resources, supervision, project administration.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The study was conducted following the World Medical Association’s Code of Ethics (Declaration of Helsinki) for experiments involving humans and was approved by the Expert Council on Ethics of the St. Petersburg State Healthcare Establishment "City Hospital No. 40" (session No. 119, February 9, 2017). Written informed consent was obtained from all subjects involved in the study.
Consent for publication
All authors agree to the publication of the manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Russkikh, I.V., Popov, O.S., Klochkova, T.G. et al. Comparative metabolomic analysis reveals shared and unique features of COVID-19 cytokine storm and surgical sepsis. Sci Rep 15, 6622 (2025). https://doi.org/10.1038/s41598-025-90426-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-90426-0
