Endocytic Pathway of Feline Coronavirus for Cell Entry: Differences in Serotype-Dependent Viral Entry Pathway



Abstract
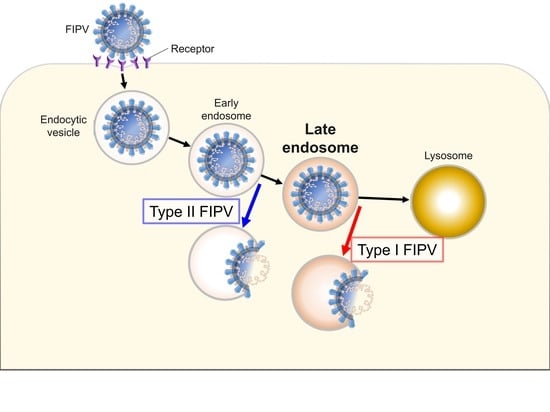
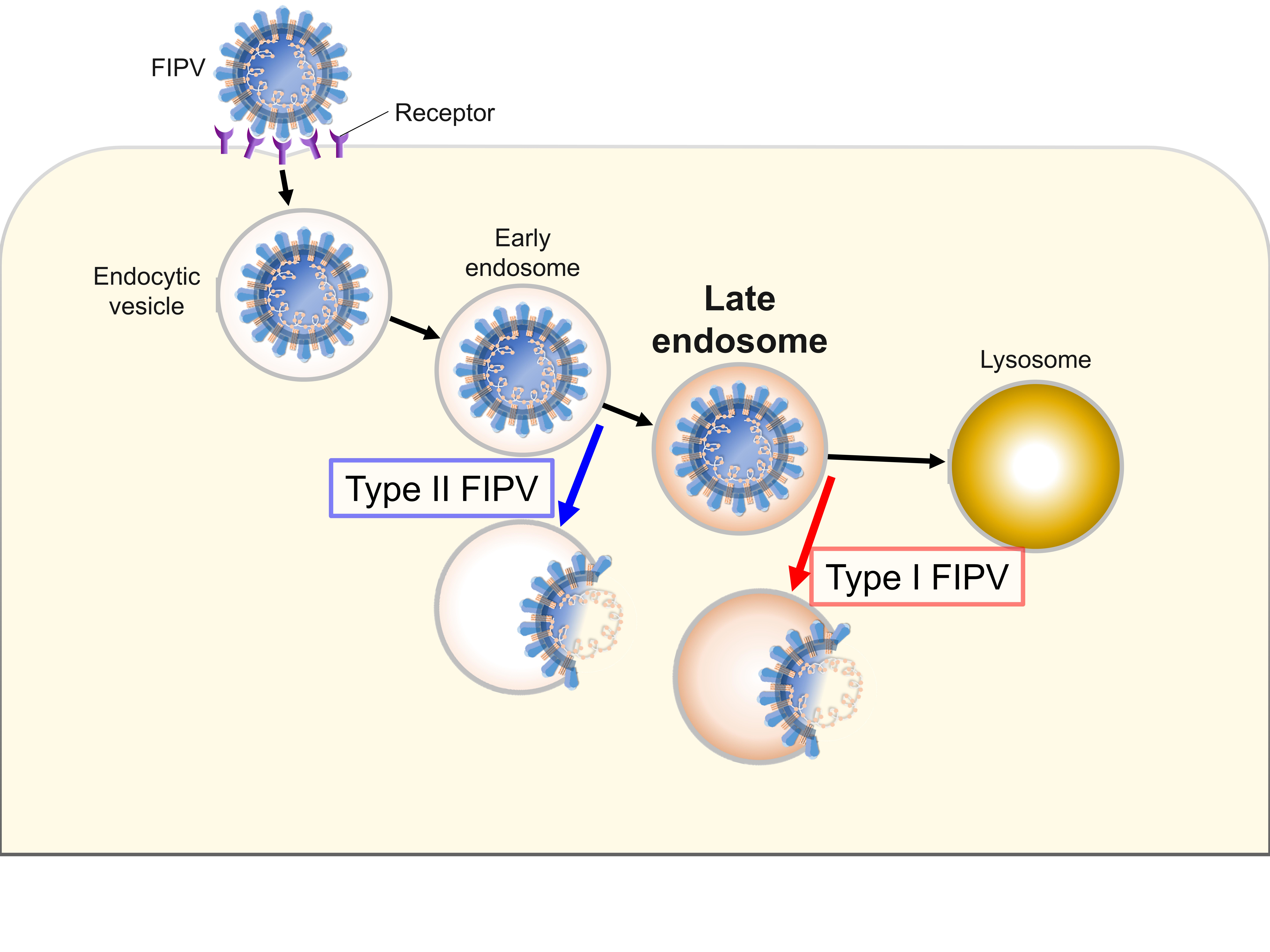
:
{kind=link}
{kind=link}
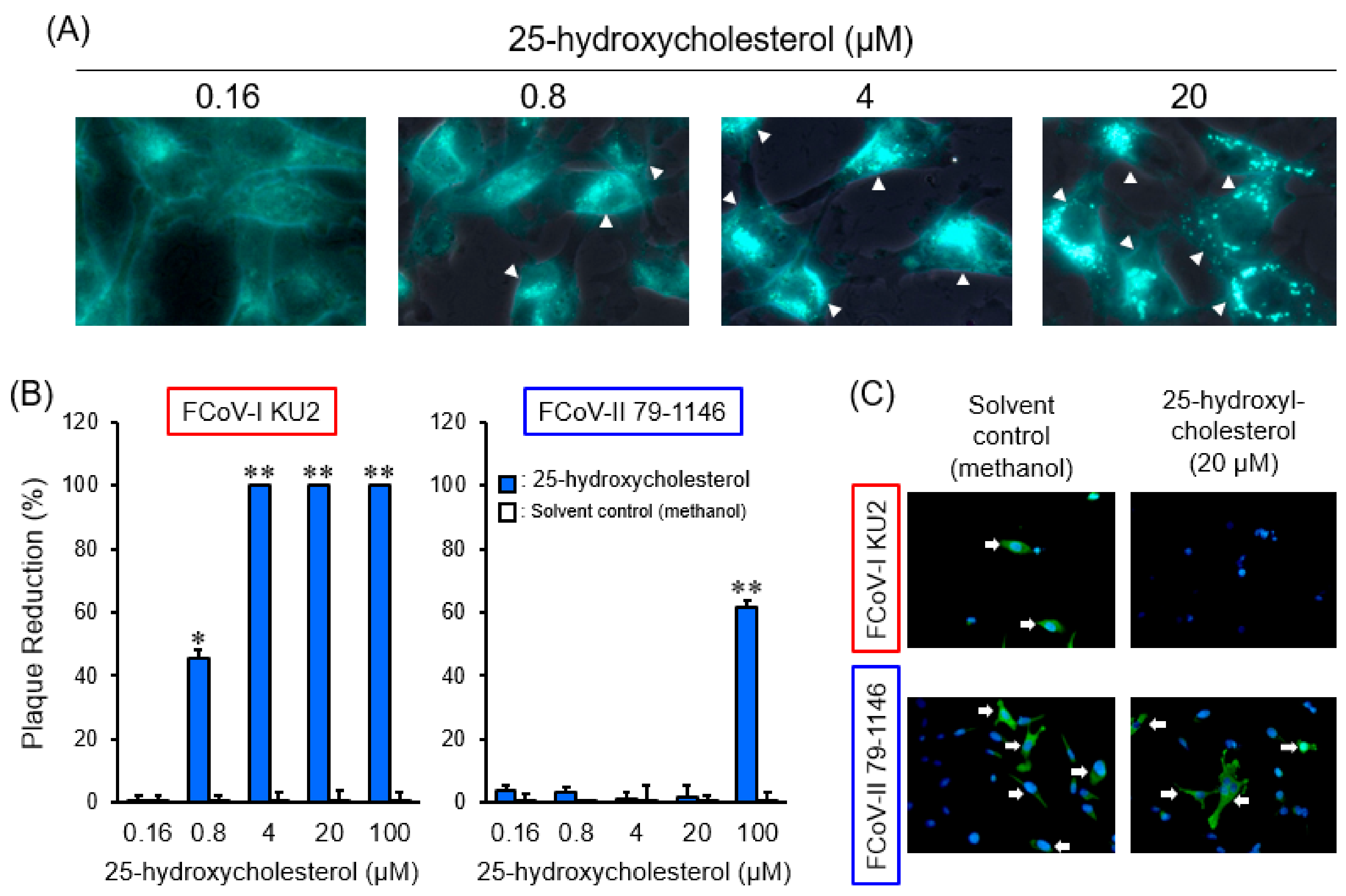{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
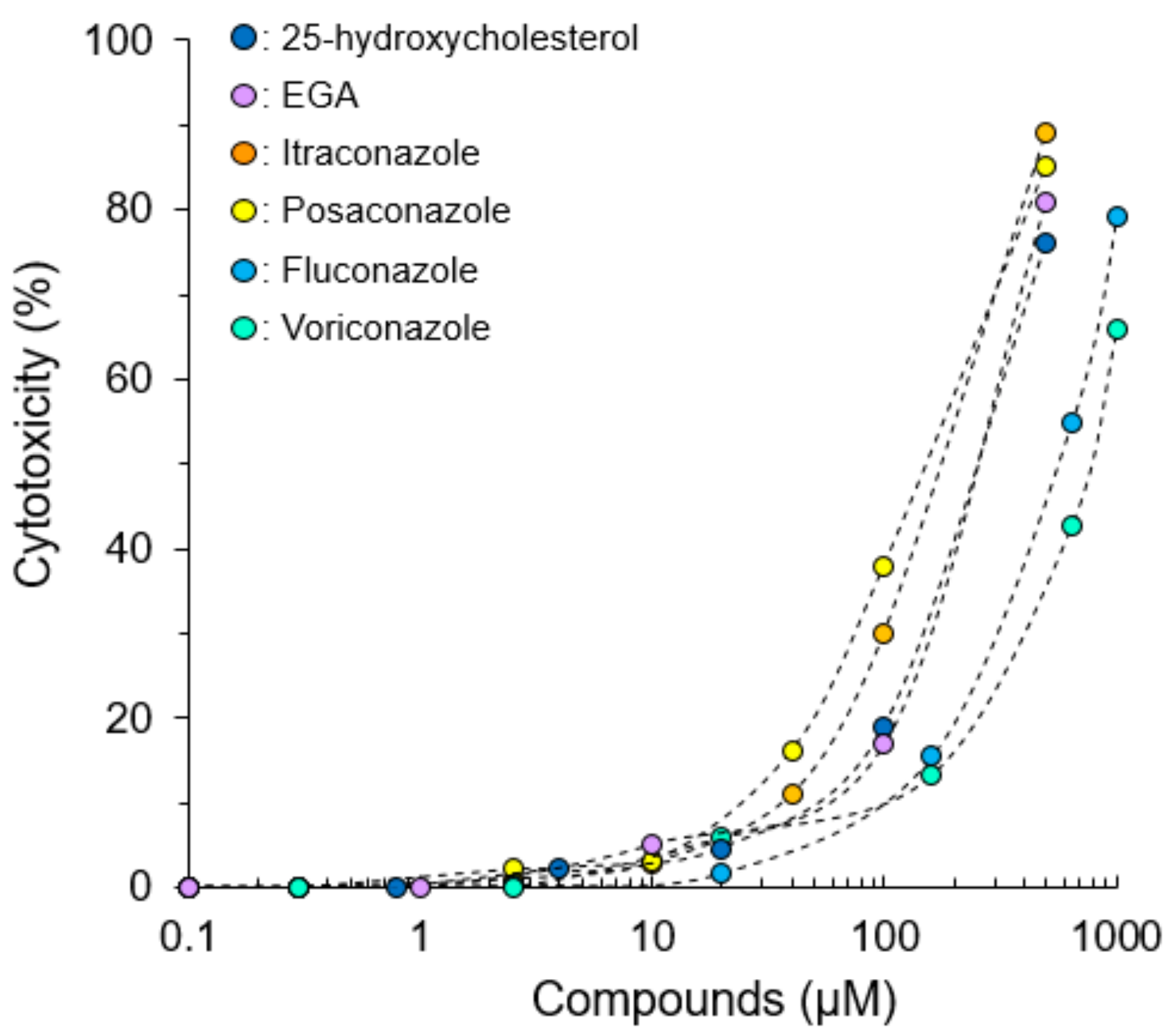
2.1. Cytotoxicity of Compounds
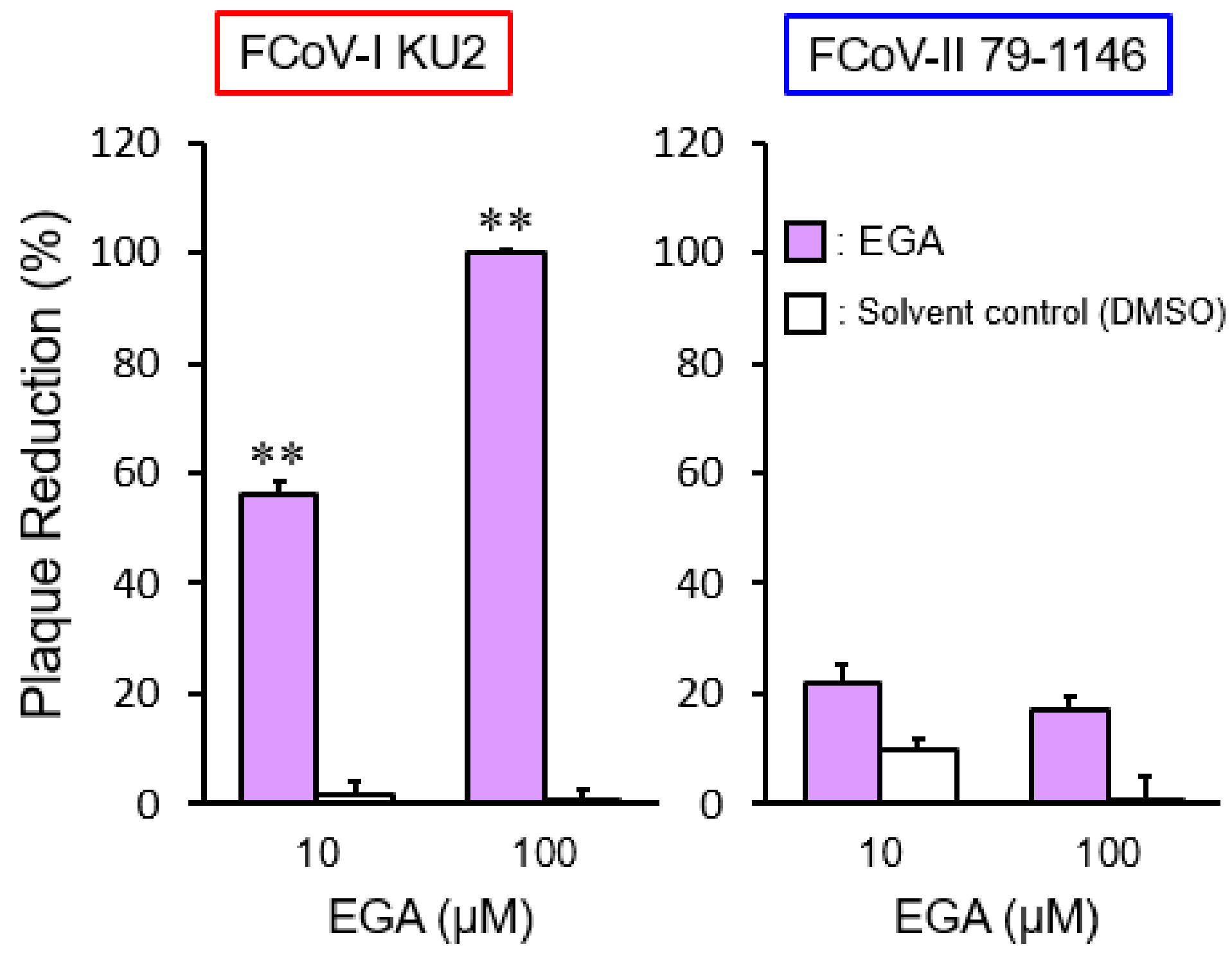
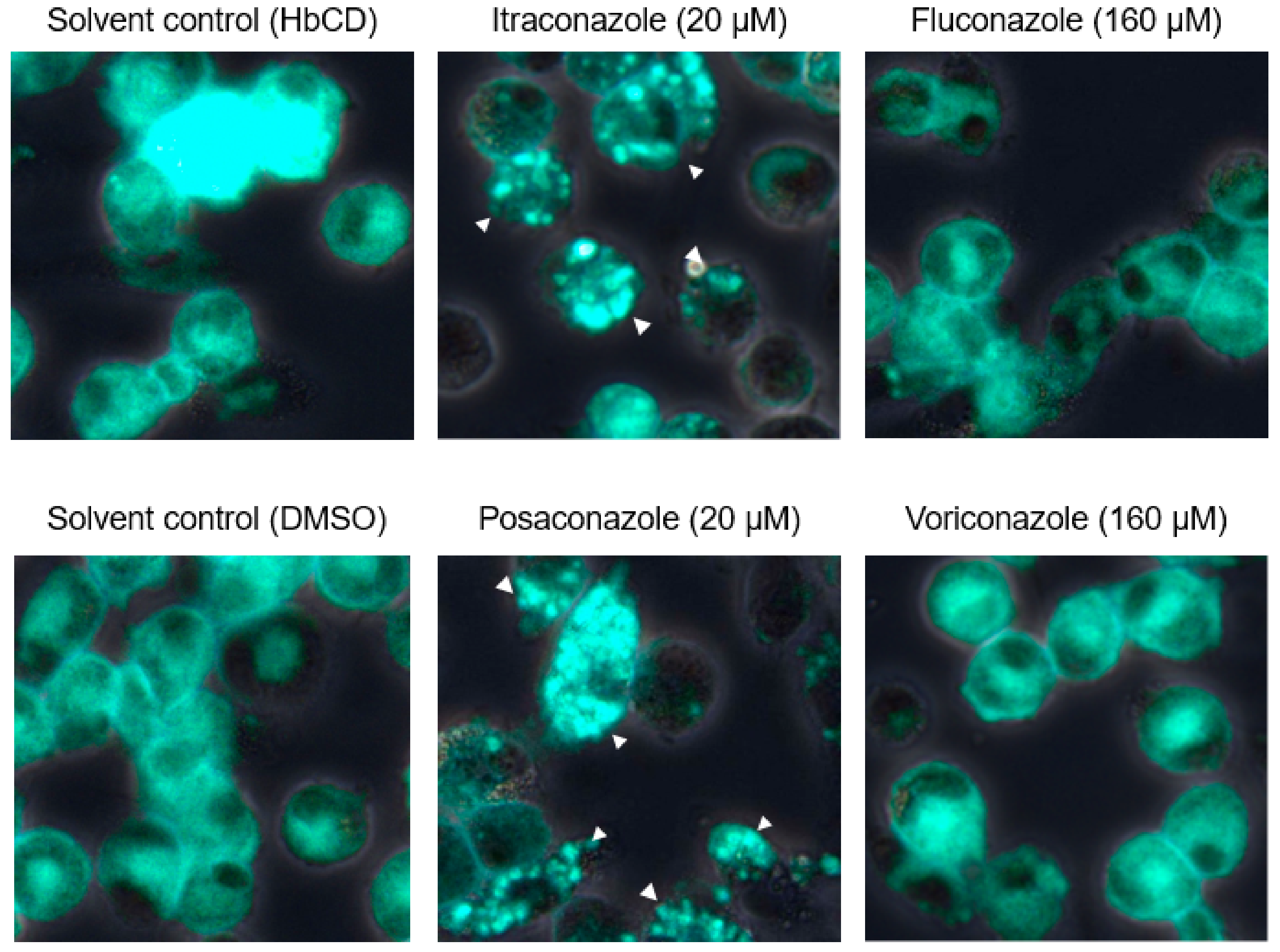
2.2. The Effects of a Late Endosomal Trafficking Inhibitor on FCoV Infection
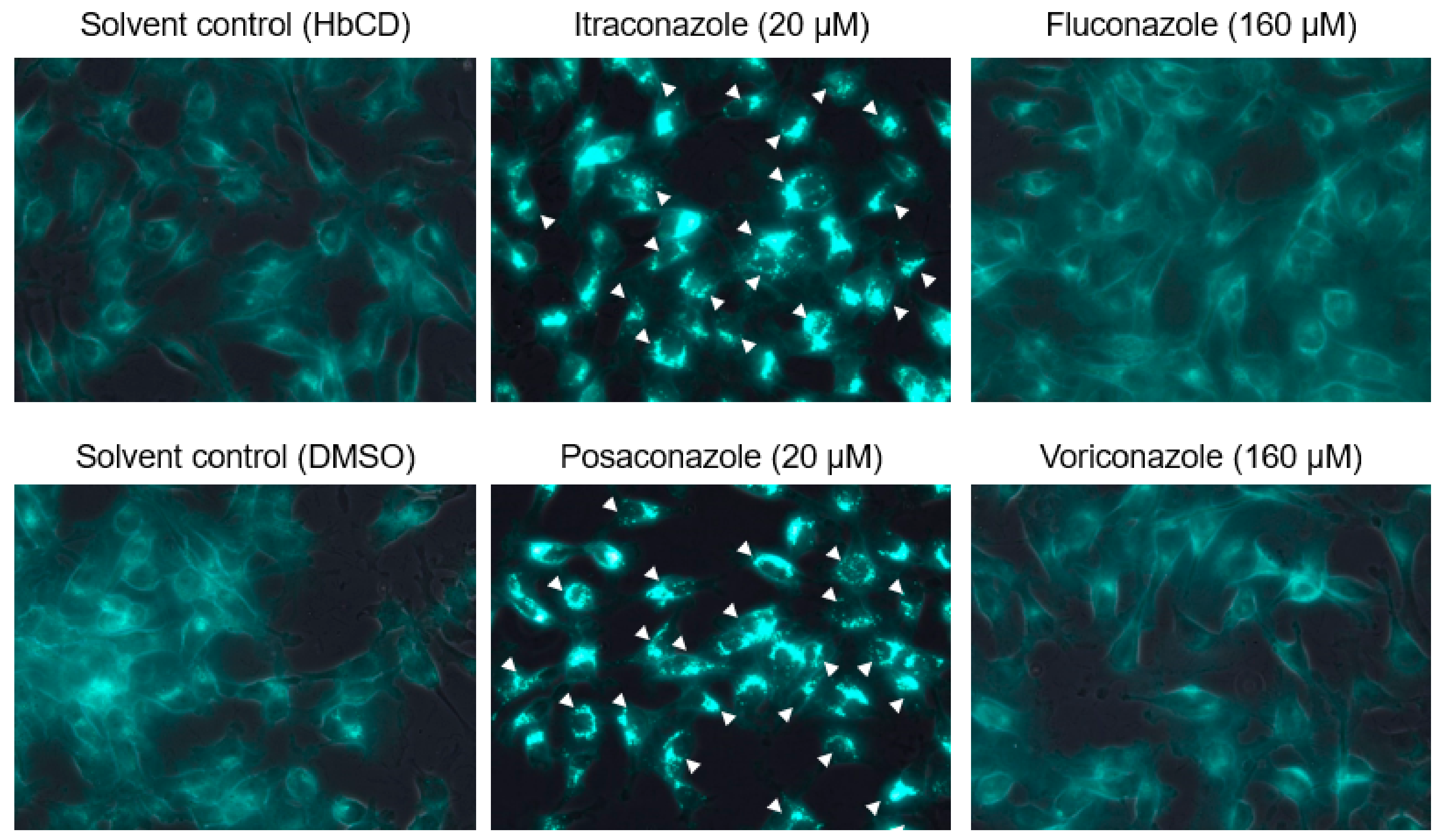
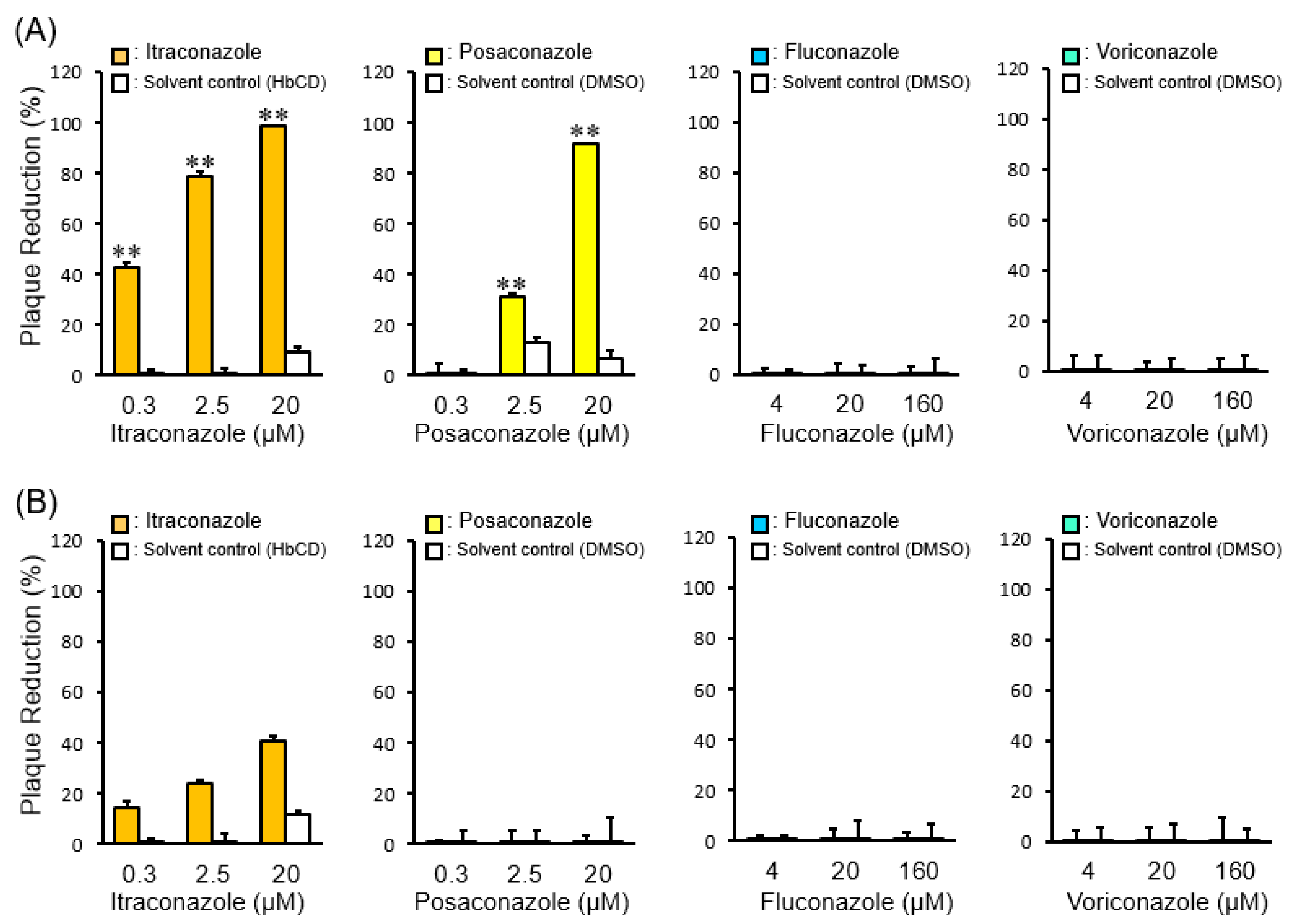
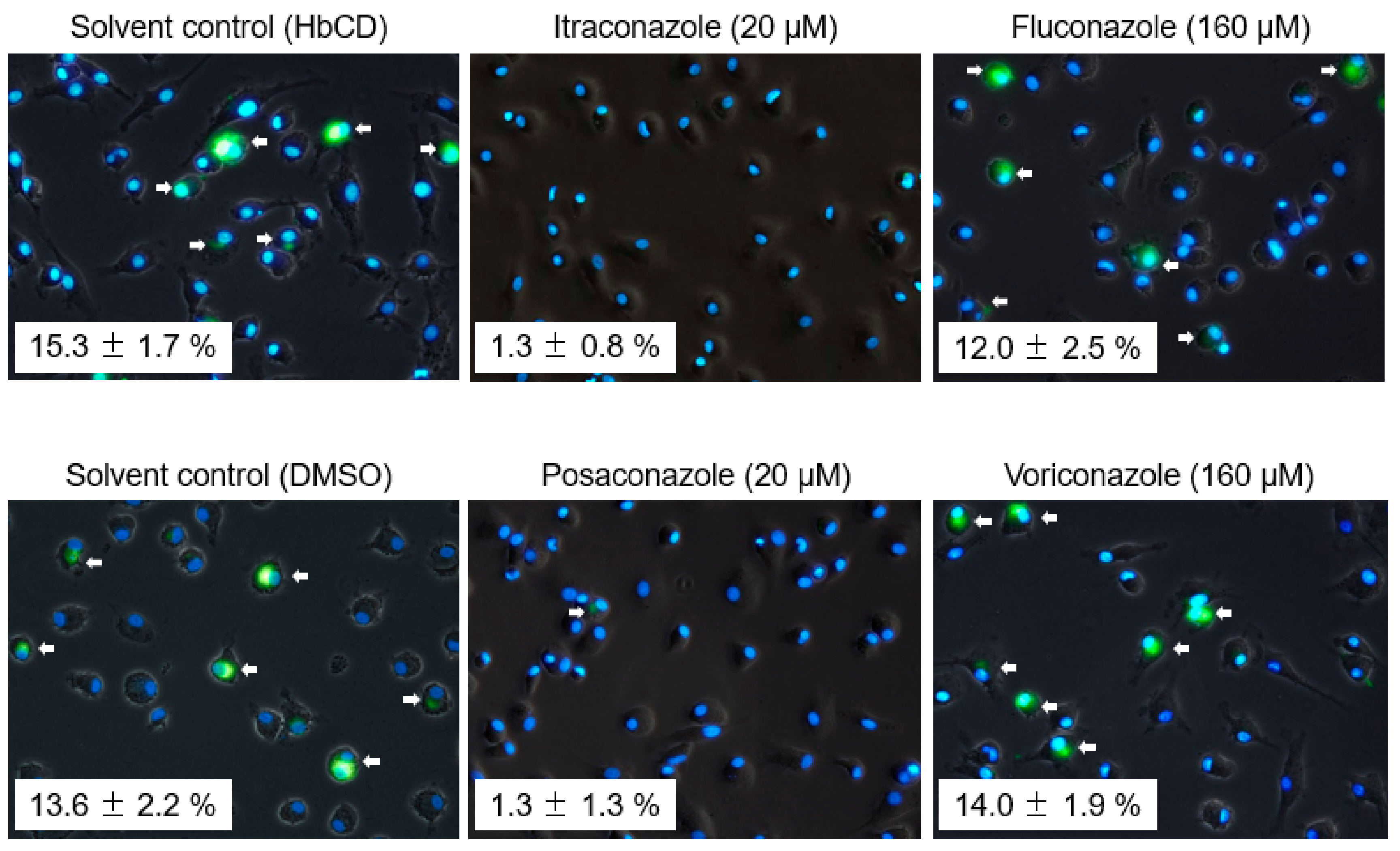
2.3. The Effects of Late Endosomal Function on FCoV in Fcwf-4 Cells
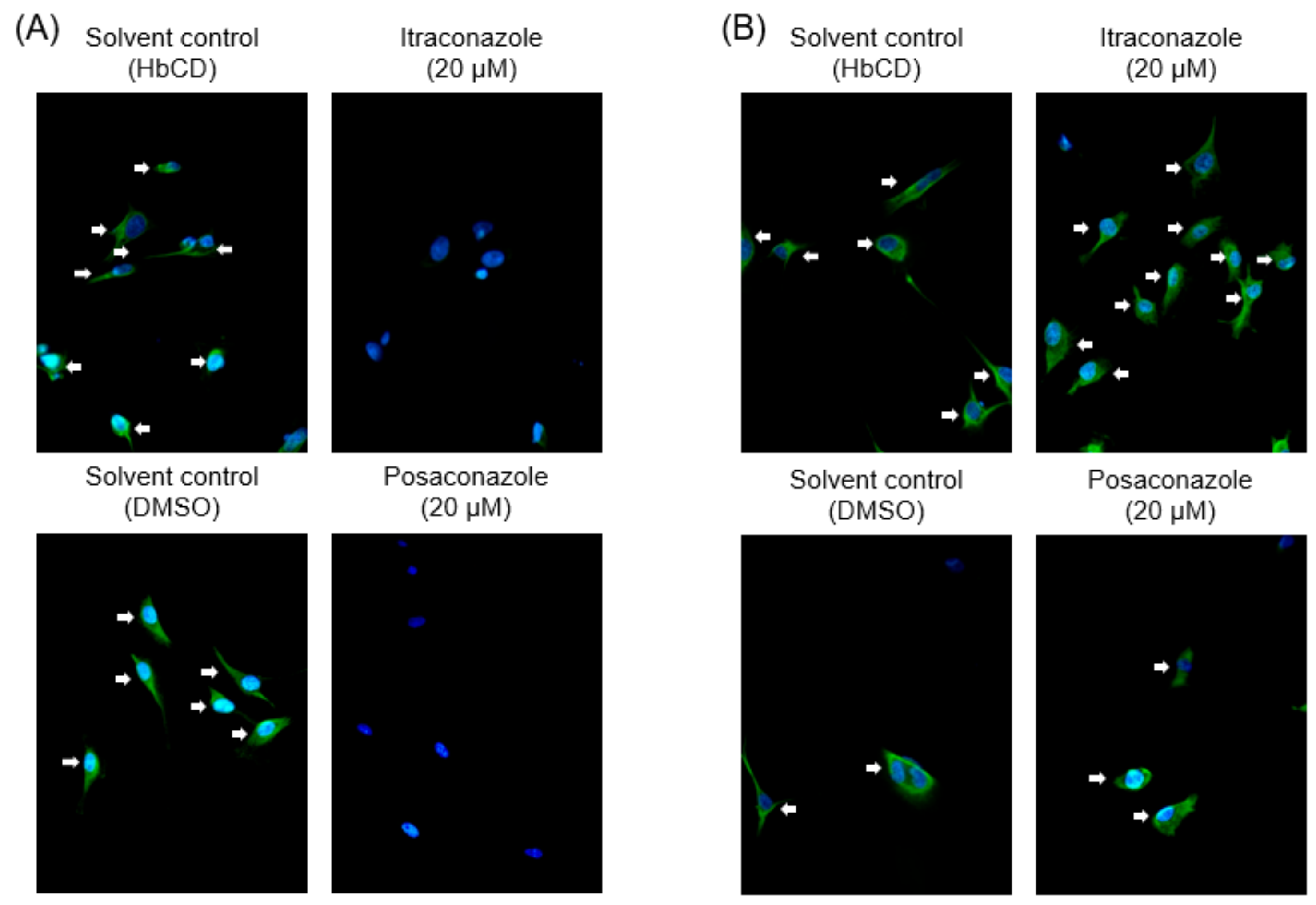
2.4. The Effects of Late Endosomal Function on FCoV in Primary Macrophages
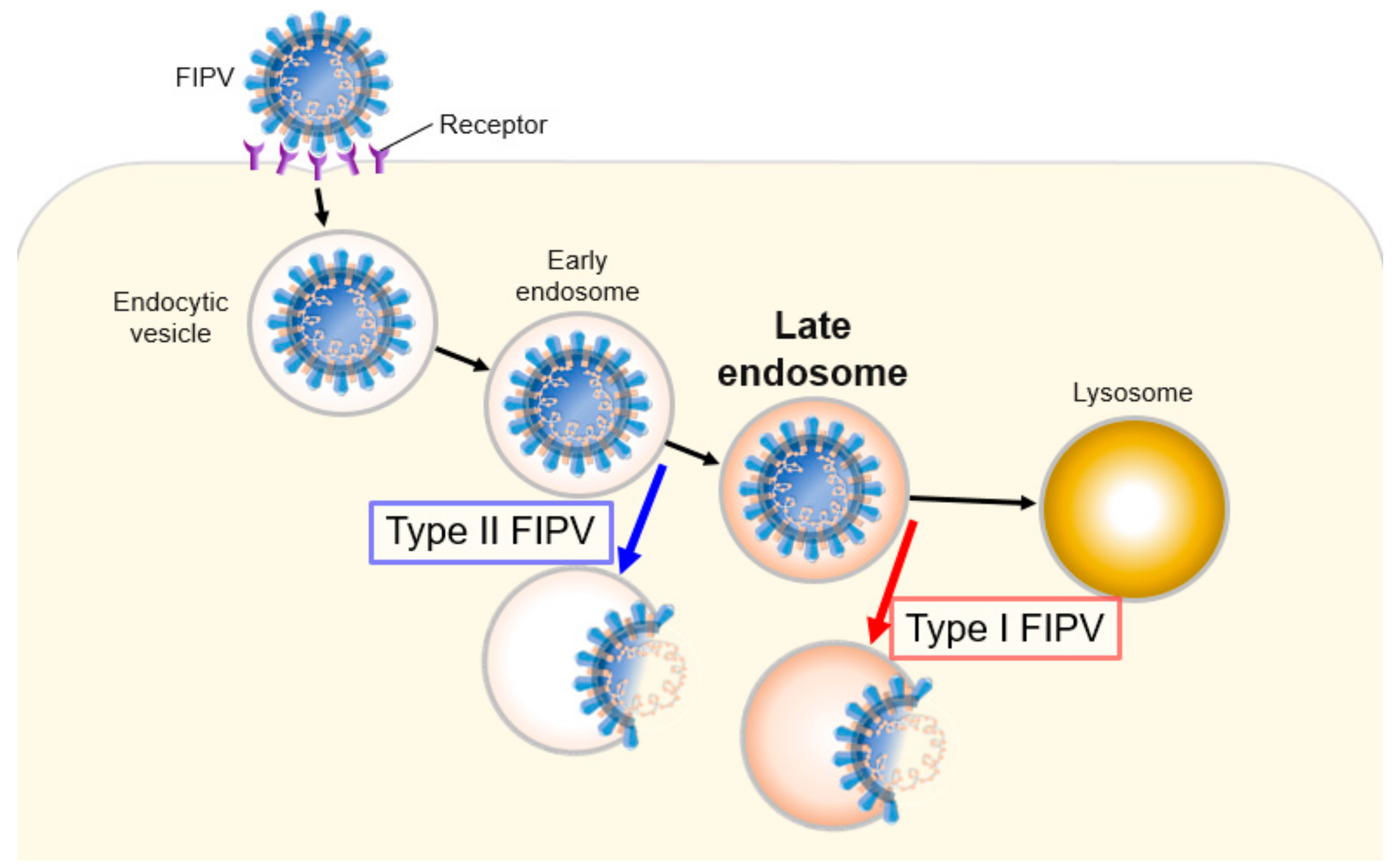
3. Discussion
4. Materials and Methods
4.1. Cell Cultures, Animals, and Viruses
4.2. Compounds
4.3. Cytotoxic Effects of Compounds
4.4. Virus Infection and Compound Treatment
4.5. Immunofluorescence Assay
4.6. Detection of Cellular Cholesterol
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Motokawa, K.; Hohdatsu, T.; Aizawa, C.; Koyama, H.; Hashimoto, H. Molecular cloning and sequence determination of the peplomer protein gene of feline infectious peritonitis virus type I. Arch. Virol. 1995, 140, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Hohdatsu, T.; Okada, S.; Ishizuka, Y.; Yamada, H.; Koyama, H. The prevalence of types I and II feline coronavirus infections in cats. J. Vet. Med. Sci. 1992, 54, 557–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummrow, M.; Meli, M.L.; Haessig, M.; Goenczi, E.; Poland, A.; Pedersen, N.C.; Hofmann-Lehmann, R.; Lutz, H. Feline coronavirus serotypes 1 and 2: Seroprevalence and association with disease in Switzerland. Clin. Diagn. Lab. Immunol. 2005, 12, 1209–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.T.; Chueh, L.L.; Wan, C.H. An eight-year epidemiologic study based on baculovirus-expressed type-specific spike proteins for the differentiation of type I and II feline coronavirus infections. BMC Vet. Res. 2014, 10, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekes, G.; Thiel, H.J. Feline coronaviruses: Pathogenesis of feline infectious peritonitis. Adv. Virus Res. 2016, 96, 193–218. [Google Scholar] [PubMed]
- Pedersen, N.C. An update on feline infectious peritonitis: Virology and immunopathogenesis. Vet. J. 2016, 201, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Stoddart, C.A.; Scott, F.W. Intrinsic resistance of feline peritoneal macrophages to coronavirus infection correlates with in vivo virulence. J. Virol. 1989, 63, 436–440. [Google Scholar]
- Takano, T.; Hohdatsu, T.; Toda, A.; Tanabe, M.; Koyama, H. TNF-alpha, produced by feline infectious peritonitis virus (FIPV)-infected macrophages, upregulates expression of type II FIPV receptor feline aminopeptidase N in feline macrophages. Virology 2007, 364, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Takano, T.; Ohyama, T.; Kokumoto, A.; Satoh, R.; Hohdatsu, T. Vascular endothelial growth factor (VEGF), produced by feline infectious peritonitis (FIP) virus-infected monocytes and macrophages, induces vascular permeability and effusion in cats with FIP. Virus Res. 2011, 158, 161–168. [Google Scholar] [CrossRef]
- Takano, T.; Endoh, M.; Fukatsu, H.; Sakurada, H.; Doki, T.; Hohdatsu, T. The cholesterol transport inhibitor U18666A inhibits type I feline coronavirus infection. Antivir. Res. 2017, 145, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Akiyama, M.; Doki, T.; Hohdatsu, T. Antiviral activity of itraconazole against type I feline coronavirus infection. Vet. Res. 2019, 50, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Dang, Y.; Ren, Y.R.; Liu, J.O. Cholesterol trafficking is required for mTOR activation in endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4764–4769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poh, M.K.; Shui, G.; Xie, X.; Shi, P.Y.; Wenk, M.R.; Gu, F. U18666A, an intra-cellular cholesterol transport inhibitor, inhibits dengue virus entry and replication. Antivir. Res. 2012, 93, 191–198. [Google Scholar] [CrossRef]
- Lyu, J.; Yang, E.J.; Shim, J.S. Cholesterol trafficking: An emerging therapeutic target for angiogenesis and cancer. Cells 2019, 8, 389. [Google Scholar] [CrossRef] [Green Version]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.C.; Farzan, M.; Jung, J.U. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Sobo, K.; Le Blanc, I.; Luyet, P.P.; Fivaz, M.; Ferguson, C.; Parton, R.G.; Gruenberg, J.; van der Goot, F.G. Late endosomal cholesterol accumulation leads to impaired intra-endosomal trafficking. PLoS ONE 2007, 2, e851. [Google Scholar] [CrossRef]
- Strating, J.R.; van der Linden, L.; Albulescu, L.; Bigay, J.; Arita, M.; Delang, L.; Leyssen, P.; van der Schaar, H.M.; Lanke, K.H.; Thibaut, H.J.; et al. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep. 2015, 10, 600–615. [Google Scholar] [CrossRef] [Green Version]
- Civra, A.; Francese, R.; Gamba, P.; Testa, G.; Cagno, V.; Poli, G.; Lembo, D. 25-Hydroxycholesterol and 27-hydroxycholesterol inhibit human rotavirus infection by sequestering viral particles into late endosomes. Redox Biol. 2018, 19, 318–330. [Google Scholar] [CrossRef]
- Gillespie, E.J.; Ho, C.L.C.; Balaji, K.; Clemens, D.L.; Deng, G.; Wang, Y.E.; Elsaesser, H.J.; Tamilselvam, B.; Gaargi, A.; Dixon, S.D.; et al. Selective inhibitor of endosomal trafficking pathways exploited by multiple toxins and viruses. Proc. Natl. Acad. Sci. USA 2013, 110, E4904–E4912. [Google Scholar] [CrossRef] [Green Version]
- Vonderheit, A.; Helenius, A. Rab7 associates with early endosomes to mediate sorting and transport of Semliki forest virus to late endosomes. PLoS Biol. 2005, 3, e233. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010, 6, e1001110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gastaldelli, M.; Imelli, N.; Boucke, K.; Amstutz, B.; Meier, O.; Greber, U.F. Infectious adenovirus type 2 transport through early but not late endosomes. Traffic 2008, 9, 2265–2278. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, Y.; Snijder, E.J.; Dobbe, J.C.; Schleich, S.; Denison, M.R.; Spaan, W.J.; Locker, J.K. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 1999, 73, 7641–7657. [Google Scholar] [PubMed]
- Quirin, K.; Eschli, B.; Scheu, I.; Poort, L.; Kartenbeck, J.; Helenius, A. Lymphocytic choriomeningitis virus uses a novel endocytic pathway for infectious entry via late endosomes. Virology 2008, 378, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Burkard, C.; Verheije, M.H.; Wicht, O.; van Kasteren, S.I.; van Kuppeveld, F.J.; Haagmans, B.L.; Pelkmans, L.; Rottier, P.J.; Bosch, B.J.; de Haan, C.A. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014, 10, e1004502. [Google Scholar] [CrossRef] [Green Version]
- Stadler, K.; Ha, H.R.; Ciminale, V.; Spirli, C.; Saletti, G.; Schiavon, M.; Bruttomesso, D.; Bigler, L.; Follath, F.; Pettenazzo, A.; et al. Amiodarone alters late endosomes and inhibits SARS coronavirus infection at a post-endosomal level. Am. J. Respir. Cell Mol. Biol. 2008, 39, 142–149. [Google Scholar] [CrossRef]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [Green Version]
- de Haan, C.A.; Stadler, K.; Godeke, G.J.; Bosch, B.J.; Rottier, P.J. Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell-cell but not virus-cell fusion. J. Virol. 2004, 78, 6048–6054. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.; Reeves, J.D.; Rennekamp, A.J.; Amberg, S.M.; Piefer, A.J.; Bates, P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. USA 2004, 101, 4240–4245. [Google Scholar] [CrossRef] [Green Version]
- Bosch, B.J.; Bartelink, W.; Rottier, P.J. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J. Virol. 2008, 82, 8887–8890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Dominguez, S.R.; Holmes, K.V. Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS-CoV) in virus entry and syncytia formation. PLoS ONE 2013, 8, e76469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, G.; Zmora, P.; Gierer, S.; Heurich, A.; Pöhlmann, S. Proteolytic activation of the SARS-coronavirus spike protein: Cutting enzymes at the cutting edge of antiviral research. Antivir. Res. 2013, 100, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Cermak, S.; Kosicek, M.; Mladenovic-Djordjevic, A.; Smiljanic, K.; Kanazir, S.; Hecimovic, S. Loss of Cathepsin B and l leads to lysosomal dysfunction, NPC-like cholesterol sequestration and accumulation of the key Alzheimer’s proteins. PLoS ONE 2016, 11, e0167428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, A.D.; Shraybman, R.; Cohen, R.D.; Whittaker, G.R. Differential role for low pH and cathepsin-mediated cleavage of the viral spike protein during entry of serotype II feline coronaviruses. Vet. Microbiol. 2008, 132, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Deng, Y.Q.; Wang, S.; Ma, F.; Aliyari, R.; Huang, X.Y.; Zhang, N.N.; Watanabe, M.; Dong, H.L.; Liu, P.; et al. 25-Hydroxycholesterol protects host against Zika virus infection and its associated microcephaly in a mouse model. Immunity 2017, 46, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Civra, A.; Cagno, V.; Donalisio, M.; Biasi, F.; Leonarduzzi, G.; Poli, G.; Lembo, D. Inhibition of pathogenic non-enveloped viruses by 25-hydroxycholesterol and 27-hydroxycholesterol. Sci. Rep. 2014, 4, 7487. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava-Ranjan, P.; Bergeron, É.; Chakrabarti, A.K.; Albariño, C.G.; Flint, M.; Nichol, S.T.; Spiropoulou, C.F. 25-Hydroxycholesterol inhibition of Lassa virus infection through aberrant GP1 glycosylation. MBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. Antiviral effect of 25-hydroxycholesterol against porcine reproductive and respiratory syndrome virus in vitro. Antivir. Ther. 2018, 23, 395–404. [Google Scholar] [CrossRef]
- Shawli, G.T.; Adeyemi, O.O.; Stonehouse, N.J.; Herod, M.R. The Oxysterol 25-Hydroxycholesterol Inhibits Replication of Murine Norovirus. Viruses 2019, 11, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Singhal, O.P. Cholesterol oxides and atherosclerosis: A review. J. Sci. Food Agric. 1991, 55, 497–510. [Google Scholar] [CrossRef]
- Shibata, N.; Glass, C.K. Macrophages, oxysterols and atherosclerosis. Circ. J. 2010, 74, 2045–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, T.; Katada, Y.; Moritoh, S.; Ogasawara, M.; Satoh, K.; Satoh, R.; Tanabe, M.; Hohdatsu, T. Analysis of the mechanism of antibody-dependent enhancement of feline infectious peritonitis virus infection: Aminopeptidase N is not important and a process of acidification of the endosome is necessary. J. Gen. Virol. 2008, 89, 1025–1029. [Google Scholar] [CrossRef]
- Trinh, M.N.; Lu, F.; Li, X.; Das, A.; Liang, Q.; De Brabander, J.K.; Brown, M.S.; Goldstein, J.L. Triazoles inhibit cholesterol export from lysosomes by binding to NPC1. Proc. Natl. Acad. Sci. USA 2017, 114, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Head, S.A.; Shi, W.Q.; Yang, E.J.; Nacev, B.A.; Hong, S.Y.; Pasunooti, K.K.; Li, R.J.; Shim, J.S.; Liu, J.O. Simultaneous targeting of NPC1 and VDAC1 by itraconazole leads to synergistic inhibition of mTOR signaling and angiogenesis. ACS Chem. Biol. 2016, 12, 174–182. [Google Scholar] [CrossRef]
- Takano, T.; Satomi, Y.; Oyama, Y.; Doki, T.; Hohdatsu, T. Differential effect of cholesterol on type I and II feline coronavirus infection. Arch. Virol. 2016, 161, 125–133. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, A.; Mettelman, R.C.; Volk, A.; André, N.M.; Whittaker, G.R.; Baker, S.C. Characterizing replication kinetics and plaque production of type I feline infectious peritonitis virus in three feline cell lines. Virology 2018, 525, 1–9. [Google Scholar] [CrossRef]
- Mettelman, R.C.; O’Brien, A.; Whittaker, G.R.; Baker, S.C. Generating and evaluating type I interferon receptor-deficient and feline TMPRSS2-expressing cells for propagating serotype I feline infectious peritonitis virus. Virology 2019, 537, 226–236. [Google Scholar] [CrossRef]
- Hohdatsu, T.; Sasamoto, S.; Koyama, H. Antigenic analysis of feline coronaviruses with monoclonal antibodies (MAbs): Preparation of MAbs which discriminate between FIPV strain 79-1146 and FECV strain 79-1683. Vet. Microbiol. 1991, 28, 13–24. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takano, T.; Wakayama, Y.; Doki, T. Endocytic Pathway of Feline Coronavirus for Cell Entry: Differences in Serotype-Dependent Viral Entry Pathway. Pathogens 2019, 8, 300. https://doi.org/10.3390/pathogens8040300
Takano T, Wakayama Y, Doki T. Endocytic Pathway of Feline Coronavirus for Cell Entry: Differences in Serotype-Dependent Viral Entry Pathway. Pathogens. 2019; 8(4):300. https://doi.org/10.3390/pathogens8040300
Chicago/Turabian StyleTakano, Tomomi, Yumeho Wakayama, and Tomoyoshi Doki. 2019. "Endocytic Pathway of Feline Coronavirus for Cell Entry: Differences in Serotype-Dependent Viral Entry Pathway" Pathogens 8, no. 4: 300. https://doi.org/10.3390/pathogens8040300
APA StyleTakano, T., Wakayama, Y., & Doki, T. (2019). Endocytic Pathway of Feline Coronavirus for Cell Entry: Differences in Serotype-Dependent Viral Entry Pathway. Pathogens, 8(4), 300. https://doi.org/10.3390/pathogens8040300