
Modelling the Transitioning of SARS-CoV-2 nsp3 and nsp4 Lumenal Regions towards a More Stable State on Complex Formation


Abstract
:1. Introduction
2. Results
2.1. Structure Prediction of the nsp3 and nsp4 Lumenal Regions
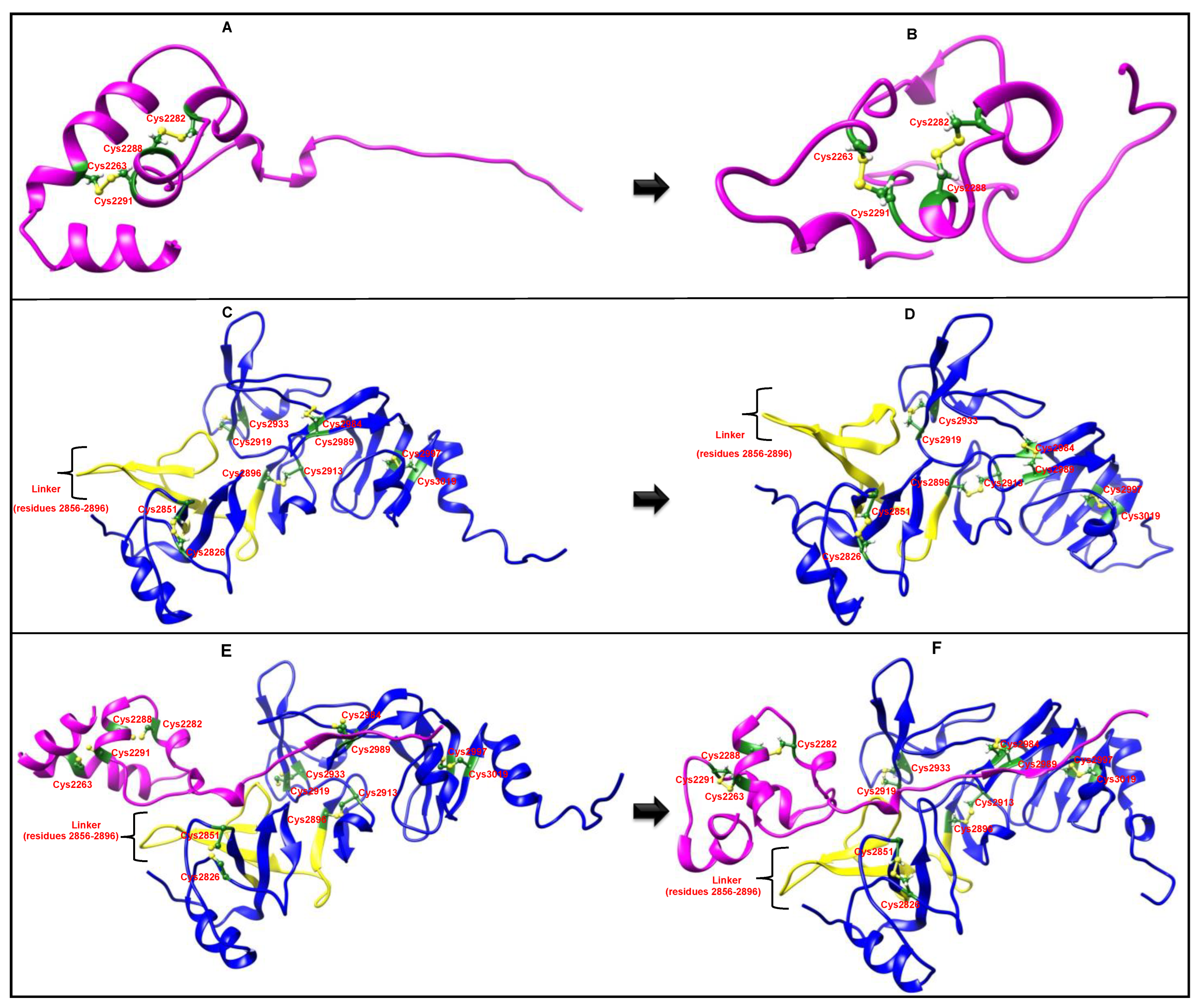
2.2. Impact of Transmembrane Regions on Modelling
2.3. MD Simulations
3. Discussion
4. Materials and Methods
4.1. Structure Prediction of the nsp3 and nsp4 Lumenal Regions
4.2. MD Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19. 11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 27 September 2022).
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication–transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet Lond. Engl. 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2021, 54, 159–163. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Serrano, P.; Johnson, M.A.; Chatterjee, A.; Neuman, B.W.; Joseph, J.S.; Buchmeier, M.J.; Kuhn, P.; Wüthrich, K. Nuclear Magnetic Resonance Structure of the Nucleic Acid-Binding Domain of Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein 3. J. Virol. 2009, 83, 12998–13008. [Google Scholar] [CrossRef] [Green Version]
- Stobart, C.C.; Sexton, N.R.; Munjal, H.; Lu, X.; Molland, K.L.; Tomar, S.; Mesecar, A.D.; Denison, M.R. Chimeric exchange of coronavirus nsp5 proteases (3CLpro) identifies common and divergent regulatory determinants of protease activity. J. Virol. 2013, 87, 12611–12618. [Google Scholar] [CrossRef] [Green Version]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef] [Green Version]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Shcherbik, N.; Sawaya, B.E. Why do SARS-CoV-2 NSPs rush to the ER? J. Neurol. 2021, 268, 2013–2022. [Google Scholar] [CrossRef]
- Sakai, Y.; Kawachi, K.; Terada, Y.; Omori, H.; Matsuura, Y.; Kamitani, W. Two-amino acids change in the nsp4 of SARS coronavirus abolishes viral replication. Virology 2017, 510, 165–174. [Google Scholar] [CrossRef]
- Gorkhali, R.; Koirala, P.; Rijal, S.; Mainali, A.; Baral, A.; Bhattarai, H.K. Structure and Function of Major SARS-CoV-2 and SARS-CoV Proteins. Bioinforma. Biol. Insights 2021, 15, 11779322211025876. [Google Scholar] [CrossRef]
- Neuman, B.W. Bioinformatics and functional analyses of coronavirus nonstructural proteins involved in the formation of replicative organelles. Antiviral Res. 2016, 135, 97–107. [Google Scholar] [CrossRef]
- Rep-Replicase Polyprotein 1ab-Severe Acute Respiratory Syndrome Coronavirus 2 (2019-nCoV)|UniProtKB|UniProt. Available online: https://www.uniprot.org/uniprotkb/P0DTD1/entry (accessed on 27 September 2022).
- UniProt Consortium UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Oostra, M.; te Lintelo, E.G.; Deijs, M.; Verheije, M.H.; Rottier, P.J.M.; de Haan, C.A.M. Localization and Membrane Topology of Coronavirus Nonstructural Protein 4: Involvement of the Early Secretory Pathway in Replication. J. Virol. 2007, 81, 12323–12336. [Google Scholar] [CrossRef] [Green Version]
- Gadlage, M.J.; Sparks, J.S.; Beachboard, D.C.; Cox, R.G.; Doyle, J.D.; Stobart, C.C.; Denison, M.R. Murine hepatitis virus nonstructural protein 4 regulates virus-induced membrane modifications and replication complex function. J. Virol. 2010, 84, 280–290. [Google Scholar] [CrossRef] [Green Version]
- Knoops, K.; Kikkert, M.; van den Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [Green Version]
- Hagemeijer, M.C.; Monastyrska, I.; Griffith, J.; van der Sluijs, P.; Voortman, J.; van Bergen en Henegouwen, P.M.; Vonk, A.M.; Rottier, P.J.M.; Reggiori, F.; de Haan, C.A.M. Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology 2014, 458–459, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Front. Phys. 2019, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jehl, P.; Manguy, J.; Shields, D.C.; Higgins, D.G.; Davey, N.E. ProViz—A web-based visualization tool to investigate the functional and evolutionary features of protein sequences. Nucleic Acids Res. 2016, 44, W11–W15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensel, Z. Predicted binding interface between coronavirus nsp3 and nsp4. bioRxiv 2022. [Google Scholar] [CrossRef]
- Vriend, G.; Krause, R.; Hekkelman, M.L.; Nielsen, J.E. The WHAT IF Web Interface. Available online: https://swift.cmbi.umcn.nl/servers/html/index.html (accessed on 15 December 2022).
- Krieger, E.; Vriend, G. YASARA View-molecular graphics for all devices-from smartphones to workstations. Bioinforma. Oxf. Engl. 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. Bioinforma. 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Bekker, H.; Berendsen, H.; Dijkstra, E.J.; Achterop, S.; Drunen, R.; van der Spoel, D.; Sijbers, A.; Keegstra, H.; Reitsma, B.; Renardus, M.K.R. Gromacs: A parallel computer for molecular dynamics simulations. Phys. Comput. 1993, 92, 252–256. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinforma. Oxf. Engl. 2013, 29, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef]
- Lemkul, J.A. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5068. [Google Scholar] [CrossRef]
- Lemkul, J.A. Lysozyme in Water. Available online: http://www.mdtutorials.com/gmx/lysozyme/index.html (accessed on 30 September 2022).
- Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| β-Strand in the Linker | β-Strand Partner |
|---|---|
| Leu 2853–Ile 2858: LIAAVI | Ile 2806–Asp 2813: IIGYKAID |
| Gly 2871–Thr 2876: GTILRT | Gly 2879–Phe 2884: GDFLHF |
| Gly 2879–Phe 2884: GDFLHF | Gly 2871–Thr 2876: GTILRT |
| Ile 2895–Tyr 2897: ICY | Leu 2902–Glu 2904: LIE |
| β-Strand in the Linker | β-Strand Partner |
|---|---|
| Leu 2853–Ile 2858: LIAAVI | Ile 2806–Asp 2813: IIGYKAID Phe 2881–Pro 2886: FLHFLP |
| Thr 2872–Arg 2875: TILR | Phe 2881–Pro 2886: FLHFLP |
| Phe 2881–Pro 2886: FLHFLP | Thr 2872–Arg 2875: TILR Leu 2853–Ile 2858: LIAAVI |
| Asn 2894–Tyr 2897: NICY | Leu 2902–Tyr 2905: LIEY |
| Interaction Type | nsp3 Lumenal Region Residue | nsp4 Lumenal Region Residue | nsp3 with TMs + nsp4 with TMs | nsp3 with TMs + nsp4 without TMs | nsp3 without TMs + nsp4 with TMs |
|---|---|---|---|---|---|
| Hydrogen bond | Asn 2275 | Arg 2875 | ✓ | ✓ | ✓ |
| Asp 2296 | Lys 2830 | ✓ | ✓ | ||
| Asp 2296 | Phe 2881 | ✓ | ✓ | ✓ | |
| Ser 2303 | Cys 2851 | ✓ | ✓ | ||
| Ser 2303 | Gly 2814 | ✓ | ✓ | ✓ | |
| Ser 2303 | Gly 2815 | ✓ | ✓ | ✓ | |
| Gln 2308 | Ile 2961 | ✓ | ✓ | ✓ | |
| Gln 2308 | Arg 2985 | ✓ | ✓ | ||
| Ile 2309 | Cys 2989 | ✓ | ✓ | ||
| Ile 2309 | Gly 2987 | ✓ | ✓ | ✓ | |
| Ionic interaction | Asp 2296 | Lys 2830 | ✓ | ✓ | ✓ |
| Pi–pi interaction | Tyr 2270 | Phe 2881 | ✓ | ||
| Cation-pi interaction | Tyr 2270 | Arg 2875 | ✓ | ✓ | ✓ |
| Hydrophobic interaction | Leu 2298 | Ile 2873 | ✓ | ✓ | ✓ |
| Structure | Average RMSD | Average Radius of Gyration | Average RMSF |
|---|---|---|---|
| nsp3 lumenal region | 9.98 Å | 13.9 Å | 5.23 Å |
| nsp4 lumenal region | 4.71 Å | 21.8 Å | 2.19 Å |
| nsp3 and nsp4 lumenal regions complex | 5.11 Å | 22.93 Å | nsp3 lumenal region: 2.4 Å |
| nsp4 lumenal region: 1.56 Å |
| Description | Uniprot Identifier | Sequence Range |
|---|---|---|
| nsp3 lumenal region | P0DTD1 | 2247–2317 |
| nsp4 lumenal region | P0DTD1 | 2797–3044 |
| nsp3 and nsp4 lumenal regions | P0DTD1 | 2247–2317; 2797–3044 |
| nsp3 lumenal region with adjoining transmembrane regions | P0DTD1 | 2226–2338 |
| nsp4 lumenal region with adjoining transmembrane regions | P0DTD1 | 2776–3065 |
| nsp3, nsp4 and nsp6 lumenal regions | P0DTD1 | 2247–2317; 2797–3044; 3656–3673 |
| nsp3 lumenal region with adjoining transmembrane regions and nsp4 lumenal region with adjoining transmembrane regions | P0DTD1 | 2226–2338; 2776–3065 |
| nsp3 lumenal region with adjoining transmembrane regions and nsp4 lumenal region | P0DTD1 | 2226–2338; 2797–3044 |
| nsp4 lumenal region with adjoining transmembrane regions and nsp3 lumenal region | P0DTD1 | 2776–3065; 2247–2317 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klatte, N.; Shields, D.C.; Agoni, C. Modelling the Transitioning of SARS-CoV-2 nsp3 and nsp4 Lumenal Regions towards a More Stable State on Complex Formation. Int. J. Mol. Sci. 2023, 24, 720. https://doi.org/10.3390/ijms24010720
Klatte N, Shields DC, Agoni C. Modelling the Transitioning of SARS-CoV-2 nsp3 and nsp4 Lumenal Regions towards a More Stable State on Complex Formation. International Journal of Molecular Sciences. 2023; 24(1):720. https://doi.org/10.3390/ijms24010720
Chicago/Turabian StyleKlatte, Nele, Denis C. Shields, and Clement Agoni. 2023. "Modelling the Transitioning of SARS-CoV-2 nsp3 and nsp4 Lumenal Regions towards a More Stable State on Complex Formation" International Journal of Molecular Sciences 24, no. 1: 720. https://doi.org/10.3390/ijms24010720