
Abstract
Understanding shared pathways and mechanisms involved in the pathogenesis of diseases like oral squamous cell carcinoma (OSCC) and COVID-19 could lead to the development of novel therapeutic strategies and diagnostic biomarkers. This study aims to predict the interactome of OSCC and COVID-19 based on salivary inflammatory proteins. Datasets for OSCC and COVID-19 were obtained from https://www.salivaryproteome.org/differential-expression and selected for differential gene expression analysis. Differential gene expression analysis was performed using log transformation and a fold change of two. Hub proteins were identified using Cytoscape and Cytohubba, and machine learning algorithms including naïve Bayes, neural networks, gradient boosting, and random forest were used to predict hub genes. Top hub genes identified included ALDH1A1, MT-CO2, SERPINC1, FGB, and TF. The random forest model achieved the highest accuracy (93%) and class accuracy (84%). The naive Bayes model had lower accuracy (63%) and class accuracy (66%), while the neural network model showed 55% accuracy and class accuracy, possibly due to data pre-processing issues. The gradient boosting model outperformed all models with an accuracy of 95% and class accuracy of 95%. Salivary proteomic interactome analysis revealed novel hub proteins as potential common biomarkers.
Similar content being viewed by others

Introduction
Saliva is a valuable source for biomarker discovery in diseases like oral squamous cell carcinoma (OSCC) and COVID-191. It offers advantages over traditional blood-based biomarker discovery, including non-invasive collection, lower risk of infections, and the ability to collect multiple samples over time to monitor disease progression or treatment response. Saliva contains SARS-CoV-2 viral RNA in infected individuals, making it a useful medium for early detection2. Changes in the salivary proteome can reflect the body’s response to SARS-CoV-2 infection, potentially serving as indicators of disease severity or progression. Non-invasive sampling allows frequent monitoring of patients, which is crucial for managing and controlling the spread of COVID-19.
Saliva contains a wide range of proteins and nucleic acids shed from the oral mucosa, making it a rich source for discovering biomarkers specific to OSCC. Proteomic analyses can identify alterations in salivary proteins that correlate with OSCC development and progression. Early detection of OSCC is critical for improving patient outcomes, and salivary biomarkers can potentially serve as non-invasive diagnostic tools for early-stage OSCC. However, challenges include standardization of collection and analysis, validation of biomarkers, and integration with other diagnostic tools3.
Saliva presents a promising source of proteins for detecting and monitoring OSCC and COVID-19, and ongoing research and technological advancements in proteomics and molecular diagnostics are likely to expand its utility further.
Hub genes in the interactome play a central role in the molecular network of interactions and pathways associated with a biological process or disease. In COVID-19, these genes are crucial for viral infection progression, host immune response, and disease severity4. ACE2 stands out as a key hub gene in the interactome, serving as the primary receptor for SARS-CoV-2, allowing the virus to enter host cells and initiate infection. Variations in ACE2 and its expression levels have been linked to differences in susceptibility to infection and the severity of COVID-19. TMPRSS2 is another important hub gene responsible for priming the viral spike protein and facilitating viral entry. Targeting TMPRSS2 has been proposed as a potential therapeutic strategy. Additionally, other hub genes in the interactome have been implicated in COVID-19, including those involved in cytokine signaling, coagulation, and cellular metabolism. Understanding these interactions is essential for identifying therapeutic targets and developing effective treatment strategies5.
OSCC constitutes most oral cancer cases, accounting for over 90% of all oral malignancies6. The disease is characterized by complex molecular alterations and dysregulated pathways. Genomic studies have identified numerous hub genes that play pivotal roles in OSCC development and progression. TP53, a tumor suppressor gene, is frequently mutated in OSCC, promoting tumor progression, invasion, and therapy resistance7. EGFR, a key gene involved in tumor growth, invasion, and angiogenesis, has been recognized as a potential therapeutic target. Other hub genes include PTGS2, CCND1, and STAT3. Understanding the functions of these genes is crucial for developing targeted therapies and identifying potential biomarkers.
Advancements in OSCC genomics can facilitate personalized treatment approaches and the discovery of novel therapeutic targets and predictive markers for prognosis and treatment response8. A recent study identified 2071 upregulated and 1893 downregulated genes in OSCC, with five hub genes (IRF4, CCR7, TNFRSF17, CD27, and S1PR4) predicted as favorable prognostic markers9. Low expression of these genes was associated with worse overall survival, while increased expression correlated with immune cell infiltration and potential therapeutic targets10,11.
The COVID-19 pandemic has had a significant impact on global health, resulting in millions of cases and considerable morbidity and mortality5. Understanding the molecular mechanisms of infection is crucial for the development of effective therapeutic strategies. Genomic studies have identified several hub genes that play central roles in the COVID-19 interactome, which are involved in essential biological processes. ACE2, TMPRSS2, immune-related genes, and genes involved in cytokine signaling, coagulation, and cellular metabolism are among the hub genes implicated in the COVID-19 interactome.
Further research into the genomics of COVID-19 and the functional characterization of these hub genes may lead to the development of targeted therapies and improved clinical management of the disease12. A previous study identified 15 potential drug targets for COVID-19 by analyzing the immune response of human cells infected with SARS-CoV-213. It highlights specific interferon-stimulated genes (ISGs) as targets, including IFIT1, IFITM1, IRF7, ISG15, MX1, and OAS2, and suggests using TLR3 agonists for drug repurposing in COVID-19 treatment, proposing modulation of the innate immune response as a valuable strategy14,15.
Network biology approaches facilitate the identification of key interactions and potential drug targets. Proteomics has gained significant attention in recent years as a powerful tool for studying the molecular mechanisms underlying various diseases16,17. Two diseases that have garnered global attention are COVID-19, caused by the novel coronavirus SARS-CoV-2, and OSCC, a malignant tumor that affects the oral cavity and has a high mortality rate. Both COVID-19 and OSCC profoundly impact the immune system, particularly inflammation and the release of inflammatory proteins.
Saliva has emerged as a valuable diagnostic medium due to its non-invasive collection, abundant protein content, and direct contact with the oral cavity, respiratory tract, and blood. Inflammatory proteins present in saliva offer crucial insights into the pathogenesis and progression of diseases. Analyzing the interactome, or the network of interactions between proteins, can provide a deeper understanding of the underlying molecular mechanisms of these diseases13,18,19.
By identifying the proteins within the interactome, we can unravel potential molecular connections between the two diseases. This research has the potential to shed light on the shared pathways and mechanisms involved in the pathogenesis of COVID-19 and OSCC, ultimately leading to the development of novel therapeutic strategies and diagnostic biomarkers.
Furthermore, comprehending the interaction between these diseases could help identify patients at higher risk of severe COVID-19 symptoms and OSCC progression, enabling targeted interventions and better patient outcomes. Few studies have explored predicting interactome genes between COVID-19 and OSCC. Thus, this study aims to predict the interactome of COVID-19 and OSCC based on salivary inflammatory proteins.
Materials and methods
Samples identification and differential gene expression analysis
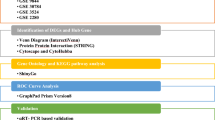
Using the dataset from COVID-19 and OSCC obtained from https://www.salivaryproteome.org/differential-expression, differential gene expression analysis was conducted. For COVID-19, the dataset IDs included 4251, 4280, 4240, and 4255, and for OSCC, the dataset IDs were 1029, 1043, 1036, and 1052. Differential gene expression analysis involved log transformation and a fold change threshold of two. This analysis is commonly used in genomics research to identify genes expressed differently between two or more conditions or groups.
The primary approach involves calculating the fold change, which quantifies the change in gene expression between conditions, and performing statistical tests such as the t-test. Before the t-test, log normalization is applied to stabilize the variance and remove systematic biases. The t-test calculates a test statistic (t-value) for each gene and converts it to a p-value. A fold change threshold of 2 is used to filter out genes with small changes, ensuring that only genes with substantial changes are considered differentially expressed. The study utilized log transformation with a fold change threshold of 2 and a P-value cutoff of 0.05 using a parametric statistical t-test. The hub gene labeled dataset underwent feature selection without needing additional normalization, and any missing data was removed through removal.
Cytoscape network and cytohubba
Cytoscape network analysis20 and hub gene identification using CytoHubba: The process involved several steps to conduct network analysis and identify hub genes using CytoHubba in Cytoscape. Firstly, gene expression data was preprocessed, followed by the construction of protein-protein interaction (PPI) networks. Cytoscape was then utilized for network visualization. Subsequently, hub genes were identified using the CytoHubba plugin, and their biological functions and potential roles were explored through functional enrichment and pathway analysis.
Machine learning of interactome hub genes
Machine learning analysis was conducted using Orange on interactome hub genes21. This process involves collecting gene expression and PPI network data, extracting relevant features, preprocessing the data, splitting it into training and testing sets, selecting a suitable machine learning algorithm, training the model, evaluating its performance using metrics, optimizing model parameters, testing the model, interpreting the results, and validating the findings. The specific implementation may vary depending on the research question, available data, and the choice of machine learning algorithms.
The preprocessing involves several steps, including data acquisition, data cleaning, outlier detection, and normalization. We adopted Data cleaning steps include removing missing values, outlier detection, and normalization, which wasn’t necessary as values are already normalized. We performed feature selection for machine learning, which involves selecting relevant features from the hub gene labelled dataset.
Random forest
Random Forest is an ensemble learning algorithm that aggregates predictions from multiple decision trees to make accurate predictions. It constructs decision trees independently using a subset of training data and features. Each decision tree is created by bootstrapping, which involves replacing samples from the original data. Additionally, a random feature subset is chosen for each tree to determine the best split at each node. Predictions are then made by combining the predictions of each tree, using the most common class in classification tasks and the average prediction in regression tasks. Random Forest employs bagging and ensemble techniques to create a robust model, reducing the risk of overfitting.
Naïve bayes
Naïve Bayes is a probabilistic classifier based on Bayes’ theorem, which follows a specific workflow and set of assumptions. It requires a labeled training dataset, where each instance contains feature values and class labels. Naive Bayes assumes that all features are conditionally independent given the class label, simplifying the calculation of class probabilities.
The classifier estimates the probability of each feature value given each class label from the training data, using probability density functions for categorical features or probability density functions for continuous features. It also estimates the prior probability of each class label from the training data. During the prediction phase, the classifier calculates the posterior probability of each class label given the feature values of a new instance.
Neural networks
Neural networks have a more intricate architecture compared to Naive Bayes, consisting of interconnected layers of neurons. This architecture, coupled with the Adam optimizer, includes the input layer, hidden layers, activation functions, and output layer. The Adam optimizer, an extension of the stochastic gradient descent algorithm, adjusts the learning rate during training by combining the advantages of AdaGrad and RMSProp optimizers.
The backpropagation algorithm calculates the difference between predicted and actual class labels through the network’s gradients. The labeled training data is divided into batches, and multiple iterations are conducted to update the model parameters using the Adam optimizer. Despite their flexibility, neural networks are more computationally demanding and necessitate larger training datasets to prevent overfitting.
Gradient boosting
Gradient boosting is a machine learning algorithm that utilizes weak learners, typically decision trees, to construct a robust predictive model. The architecture involves sequentially adding weak learners to the model ensemble, with each learner focused on correcting the mistakes of the previous ones. Residuals from the previous models are calculated, and the next weak learner is trained on these residuals to minimize errors. Predictions from each weak learner are combined to produce a final prediction, with weights determined based on their performance on the training data.
Gradient boosting also includes a learning rate parameter to regulate the contribution of each weak learner to the final prediction. Regularization techniques are commonly employed to prevent overfitting and enhance generalization.
Results
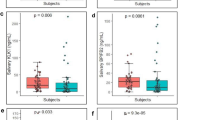
Normalized values in COVID and OSCC samples adjust for variations in total RNA or cDNA amounts, ensuring accurate comparisons (Fig. 1). Log normalization converts raw expression values into a logarithmic scale, stabilizing variance and rendering them suitable for statistical analysis. This technique is particularly beneficial for analyzing high-throughput gene expression data from microarrays or RNA sequencing technologies.

Normalized values in COVID and OSCC samples.

Volcano plot of differential gene expression.
The volcano plot illustrates the differential gene expression between COVID and OSCC samples, highlighting up-and-down-regulated genes (Fig. 2). Each point represents a gene, with the x-axis representing the log2 fold change and the y-axis representing the negative log10 of the adjusted p-value. Up-regulated genes are depicted in red, while down-regulated genes are shown in blue. This analysis provides insights into the molecular mechanisms and pathways underlying COVID and OSCC, aiding in understanding the biological processes contributing to these conditions.

Principal component analysis (PCA).
The Principal Component Analysis (PCA) plot illustrates the simplification and visualization of complex datasets (Fig. 3). In this example, PCA generated two principal components (PCs): PC-1 and PC-2. PC-1, representing 46.6% of the variance, captures the most significant patterns or trends in the data, explaining a considerable portion of the overall variability. PC-2, with a variance of -10.7%, explains a smaller variance, indicating it may represent noise or unimportant variation. These results suggest that additional principal components may be necessary to explain the remaining variance in the data.

Heat map of genomic samples.
The heat map illustrates the distribution of genomic samples for salivary COVID and OSCC, facilitating the identification of shared genetic characteristics and patterns (Fig. 4). This visualization enables quick comparisons of genomic profiles, offering insights into the genetic distribution within the dataset and potentially leading to discoveries or targeted interventions.

Interactome network of differentially expressed genes.
The interactome network illustrates the connections between differentially expressed genes in COVID and OSCC, with hub genes identified using the CytoHubba tool (Fig. 5). These hub genes are crucial for connectivity and molecular mechanisms. Notable hub genes include ALDH1A1, MT-CO2, SERPINC1, FGB, and TF.

Gene ontology and KEGG pathway analysis. (a) Cellular component: this section of the figure displays the cellular components associated with the analyzed genes. (b) Biological Processes: The biological processes related to the analyzed genes are depicted in this section. (c) Molecular Function: This section shows the molecular functions of the analyzed genes. (d) KEGG Pathway: The KEGG Pathway section highlights the Complement and Coagulation Cascades pathway, which is critical for COVID-19 and OSCC.
Dysregulation of these cascades in COVID-19 can lead to inflammation and a hypercoagulable state (Fig. 6). Similarly, in OSCC, these pathways are involved in tumor progression and immune evasion. Understanding these cascades is essential for developing targeted treatments for both diseases.
Table 1 compares the performance of machine learning models, including random forest, naive Bayes, neural networks, and gradient boosting. The random forest model achieved the highest accuracy at 93% and a class accuracy of 84%. Naive Bayes had lower accuracy at 63% and a class accuracy of 66%. The neural network model showed 55% accuracy and a class accuracy of 55%, possibly due to data preprocessing issues. Gradient boosting outperformed all models with an accuracy of 95% and a class accuracy of 95%.

ROC Curve of the Hub with multiple curves with AUC values labeled as 0.501 (cyan curve), 0.516 (orange curve), and 0.339 (purple curve). The cyan and orange curves show weak predictive power, while the purple curve has the lowest AUC (0.339). The purple curve indicates poor performance, worse than random guessing.Models with lower AUC values are less reliable for biomarker identification and indicates weaker models overall, with no curve achieving strong predictive performance.
The ROC curve illustrates the Receiver Operating Characteristic of the hub (Fig. 7).

ROC Curve of Non-Hub Genes with purple curve (AUC = 0.661) shows the best performance, indicating better sensitivity and specificity. The purple curve represents a model with moderate predictive power, while the cyan and orange curves have AUC values closer to 0.5, indicating models with performance close to random guessing.Models with higher AUC values are better at identifying true hub biomarkers while minimizing false positives. model with moderate predictive power (AUC = 0.661), which could be useful for biomarker identification.
The ROC curve depicts the Receiver Operating Characteristic of non-hub genes (Fig. 8).
The Receiver Operating Characteristic Curve (ROC) visually displays the performance of a binary classification model by plotting the true positive rate (TPR) against the false positive rate (FPR) across various classification thresholds. An ideal model achieves a TPR of 1 and an FPR of 0, forming a perfect curve, while a random model produces a diagonal line. The area under the ROC curve (AUC) serves as a metric for evaluating model performance, with higher AUC values indicating better predictive capability. The AUC is higher for models like random forest and gradient boosting, indicating greater predictive accuracy, and lower for naive Bayes and neural networks.

Confusion matrix of all algorithms.
Figure 9 displays the confusion matrix of all algorithms used in the study.
A confusion matrix is a table that summarizes a classification model’s performance on test data, showing the proportion of true positive (TP), true negative (TN), false positive (FP), and false negative (FN) predictions. It allows for calculating performance metrics like accuracy, precision, recall, specificity, and F1 score. The confusion matrix helps understand the model’s performance, particularly in balancing identification with avoiding misclassification, especially in imbalanced datasets with uneven class distribution.

Lift curve for predicting hub genes.
Figure 10 presents the lift curve, a visualization tool utilized to assess the performance of predictive models, particularly for predicting hub genes. The lift curve illustrates that random forest and gradient boosting algorithms effectively identify hub genes with high lift, which holds significance in biological applications. However, each algorithm has its strengths and weaknesses, necessitating further analysis and comparison of performance metrics to gain a comprehensive understanding of their effectiveness in this context.

Lift curve for non-hub genes.
Figure 11 displays the lift curve for all non-hub genes. The lift curve is a visualization tool used to evaluate the performance of predictive models. It demonstrates the ability of the models to accurately predict non-hub genes. Analyzing the lift curve helps in understanding the effectiveness of different algorithms in identifying non-hub genes.
Discussion
Saliva emerges as a promising source of biomarkers for medical research and diagnostics, containing various proteins such as enzymes, antibacterial compounds, growth factors, hormones, and cytokines22,23. Its non-invasive nature, ease of collection, cost-effectiveness, stability, and dynamic properties make it valuable for providing real-time insights into oral and systemic diseases23. Saliva-based biomarker research is gaining momentum in fields like oncology, infectious diseases, autoimmune diseases, metabolic disorders, and oral health. However, challenges including variability in saliva composition, potential interference from oral bacteria, dilution factors, and the need for standardized collection methods need to be addressed1,24. Further research is necessary to fully harness saliva’s potential in clinical practice22,23.
Interactome hub genes (Figs. 1, 2, 3, 4 and 5), which are central nodes in molecular interaction networks, play crucial roles in cellular processes and pathways, providing valuable insights into the underlying mechanisms of oral cancer and COVID-1925. For instance, TP53, a tumor suppressor gene, is associated with tumor progression and poor prognosis in oral cancer, while EGFR regulates innate immune responses and inflammatory processes in both diseases. Additionally, genes like STAT3, CASP3, and IL6 regulate immune responses and cell survival.
The complement and coagulation cascades, crucial in COVID-19, impact blood coagulation and inflammation through proteins like C3 and fibrinogen. Similarly, in OSCC, these cascades contribute to tumor progression and immune evasion by regulating blood coagulation and complement activation. Understanding their gene ontology and KEGG pathways offers insight into potential therapeutic targets for both conditions, influencing inflammation, immune responses, and tumor microenvironments (Fig. 6). Furthermore, comprehending these interactions may lead to targeted therapies and biomarkers, potentially repurposing existing drugs or developing new therapeutic strategies. However, further research is necessary to fully understand their complex interactions and implications25.
In this study, we identified several hub proteins and discussed their roles. ALDH1A1 is a gene encoding the enzyme ALDH1A114,15,26, which plays a crucial role in aldehyde metabolism. It is upregulated in the lung tissues of COVID-19 patients, likely due to oxidative stress. ALDH1A1 has also been identified as a potential biomarker for COVID-19 severity, suggesting its potential for predicting disease progression. In oral cancer, ALDH1A1 is a critical marker associated with cancer stem cells (CSCs), contributing to tumor initiation, progression, and therapy resistance19. The pathway of aldehyde dehydrogenase 1A1 (ALDH1A1), a marker of cancer stem-like cells (CSCs), is crucial for maintaining their properties. Studies have shown that high ALDH1A1 expression is an adverse prognostic factor in OSCC patients. Overexpression activates the AKT-β-catenin signaling pathway, while low expression enhances chemo-resistance ability and sporogenesis. ALDH, a crucial protein in the body, is regulated by the Wnt/β-catenin pathway, which depends on β-catenin and TCF, affecting its activity.
Recent evidence showed that High expression levels of ALDH1A1 in oral squamous cell carcinoma are linked to advanced TNM tumor and nodal stages, as well as increased mortality rates, establishing it as a significant marker of invasiveness and poor prognosis27,28. Patients with adenocarcinoma with SARS-CoV2 showed more cancer stem cells (CSCs) and increased lymph node metastasis cases. SARS-CoV2 proteins, Nucleocapsid and Spike, were found in lung adenocarcinoma cells, suggesting the virus may stay in the body longer and increase cancer spread also substantiaing our study results29.
Targeting ALDH1A1 in oral cancer cells has yielded promising results, reducing cancer stem cell (CSC) properties, and increasing therapy sensitivity. MT-CO2 (Mitochondrially Encoded Cytochrome C Oxidase II) is a gene encoding a subunit of cytochrome c oxidase, an essential enzyme involved in the electron transport chain in mitochondria. This enzyme plays a crucial role in cellular respiration by facilitating the final step of the energy production process. While the direct relationship between MT-CO2 and COVID-19 or oral cancer is not well-established, mitochondrial dysfunction has been implicated in the pathogenesis of these diseases. COVID-19 can cause mitochondrial damage19, impairing energy production, increasing oxidative stress, and promoting the release of pro-inflammatory cytokines. This dysfunction contributes to the severity of symptoms and complications, such as organ failure. In oral cancer, alterations in mitochondrial function result in increased reactive oxygen species (ROS) production and altered energy metabolism, promoting tumor growth, invasion, and therapy resistance. SERPINC1, a gene encoding antithrombin, has been associated with abnormal blood clotting and coagulation disorders in severe cases of the disease. Oral cancer is also associated with an increased risk of thromboembolic events. Elevated levels of fibrinogen have been associated with worse prognosis and increased risk of thrombosis in COVID-19 patients. Tissue Factor30, a gene activating clotting factors and blood clot formation, has been upregulated in COVID-19 cases, contributing to hypercoagulable states.
The KEGG pathway analysis highlights the role of complement and coagulation cascades in understanding COVID-19 and oral squamous cell carcinoma (OSCC). The activation of the Wnt/β-catenin signaling pathway, which plays a crucial role in cellular proliferation and differentiation, may contribute to OSCC malignancy. Targeting this pathway could offer a strategic therapeutic approach. Aldehyde dehydrogenase regulation may promote cancer stem cells, linking these pathways to tumor aggressiveness. Inflammatory pathways, particularly those associated with salivary proteins, suggest immune response involvement in both conditions. Coagulation dysregulation through the SERPINC1 gene may be linked to cancer progression. Metabolic alterations mediated by MT-CO2 could provide insights into metabolic shifts. Further research is needed to fully understand the specific mechanisms and implications of these genes in COVID-19 and oral cancer.
In a previous study, 257 susceptibility genes associated with RNA splicing, mitochondrial functions, and proteasomes were identified in lung adenocarcinoma (LUAD) patients with COVID-1931. Ten hub genes (MEA1, MRPL24, PPIH, EBNA1BP2, MRTO4, RABEPK, TRMT112, PFDN2, PFDN6, and NDUFS3) were significantly correlated with immune cell infiltration. SARS-CoV-2 has a higher chance of progression in adults with underlying health conditions such as cancer and neurological diseases, leading to comorbidities and potential death. Understanding host-virus protein-protein interactions is crucial for designing effective antiviral drugs for SARS-CoV-2. Identifying hub proteins and their interactions with SARS-CoV-2 proteins, which trigger comorbidities like Alzheimer’s, Parkinson’s, and prostate cancer, is essential18.
This study evaluated the performance of various machine learning algorithms in accurately predicting hub genes in OSCC and COVID-19 (Table 1). The random forest model demonstrated the highest accuracy at 93% and class accuracy at 84% (Figs. 5, 6, 7, 8, 9 and 10)32,33. In contrast, the naive Bayes model exhibited lower accuracy at 63% and class accuracy at 66%, attributed to its assumption of feature independence. The neural network model achieved an accuracy of 55% and class accuracy33, with performance heavily reliant on proper data preprocessing. However, the gradient boosting model outperformed all other models with an accuracy of 95% and class accuracy, indicating its effectiveness in learning underlying data patterns.
Predicting interactomic hub proteins using machine learning faces several challenges. One major obstacle is the availability of high-quality training data, necessitating the collection of comprehensive and diverse datasets. Generalizing across different conditions is essential, as studies often focus on specific cell types or biological conditions. Moreover, biological interpretation and validation are crucial for accurate predictions. Integrating multi-omics data, such as transcriptomics, proteomics, and epigenomics, can provide a more comprehensive understanding of molecular interactions34,35,36.
Optimizing preprocessing steps, such as data normalization and feature selection, can enhance model performance. However, interpretability remains a concern, particularly for models like random forest and gradient boosting, which are often considered black-box models36,37,38,39. Finally, incorporating network topology information can enhance the accuracy and applicability of hub gene prediction models in understanding complex biological systems.
Conclusion
The study suggests that predicting interactomic hub genes for COVID-19 and OSCC holds promise, but several challenges need to be addressed. Random forest and gradient boosting models demonstrated superior performance, while naive Bayes performed poorly.
Data availability
The datasets generated and/or analysed during the current study are available in https://www.salivaryproteome.org/differential-expression.
References
Bajaj, N., Granwehr, B. P., Hanna, E. Y. & Chambers, M. S. Salivary detection of SARS-CoV-2 (COVID-19) and implications for oral health-care providers. Head Neck. 42, 1543–1547 (2020).
Baghizadeh Fini, M. Oral saliva and COVID-19. Oral Oncol. 108, 104821 (2020).
Boutros, J. et al. Salivary detection of COVID-19: clinical performance of oral sponge sampling for SARS-CoV-2 testing. ERJ Open. Res. 7, 00396–2021 (2021).
Zhang, W., Yang, Z., Zhou, F., Wei, Y. & Ma, X. Network Pharmacology and Bioinformatics Analysis Identifies Potential Therapeutic Targets of Paxlovid Against LUAD/COVID-19. Front. Endocrinol. (Lausanne). 13, 935906 (2022).
Goldman, J. D., Gonzalez, M. A., Rüthrich, M. M., Sharon, E. & von Lilienfeld-Toal, M. COVID-19 and Cancer: special considerations for patients receiving immunotherapy and immunosuppressive Cancer therapies. Am. Soc. Clin. Oncol. Educ. Book. 42, 1–13 (2022).
Zahra, A. et al. Meta-analysis of miRNAs and their involvement as biomarkers in oral cancers. Biomed. Res. Int. 2018, 8439820 (2018).
Network, C. G. A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature [Internet]. 517(7536), 576–82 (2015). https://pubmed.ncbi.nlm.nih.gov/25631445
Lee, J. et al. Human primary epithelial cells acquire an epithelial-mesenchymal-transition phenotype during long-term infection by the oral opportunistic Pathogen, Porphyromonas gingivalis. Front. Cell. Infect. Microbiol. 7, 493 (2017).
Chujan, S., Kitkumthorn, N. & Satayavivad, J. Identification of potential molecular mechanisms and prognostic markers for oral squamous cell carcinoma: a Bioinformatics Analysis. J. Int. Soc. Prev. Community Dent. 13, 237–246 (2023).
Feller, L. L., Khammissa, R. R., Kramer, B. B. & Lemmer, J. J. Oral squamous cell carcinoma in relation to field precancerisation: pathobiology. Cancer Cell. Int. 13, 31 (2013).
Whitmore, S. E. & Lamont, R. J. Oral bacteria and cancer. PLoS Pathog. 10, e1003933 (2014).
Lyons, A., Mcdonald, C., Kanatas, A. & Rogers, S. N. Early oral cancer management during the COVID-19 period. Br. J. Oral Maxillofac. Surg. 58, 885–887 (2020).
Osuna-Martinez, U. et al. In Silico Prediction of hub genes involved in Diabetic kidney and COVID-19 related disease by Differential Gene expression and Interactome Analysis. Genes (Basel). 13, 2412 (2022).
Pathania, A. S. et al. COVID-19 and Cancer Comorbidity: Therapeutic opportunities and challenges. Theranostics 11, 731–753 (2021).
Monk, B. J. et al. COVID-19 and ovarian cancer: exploring alternatives to intravenous (IV) therapies. Gynecol. Oncol. 158, 34–36 (2020).
Yadalam, P. K. et al. Gene and protein interaction network analysis in the epithelial-mesenchymal transition of Hertwig’s epithelial Root Sheath reveals periodontal regenerative drug targets – an in silico study. Saudi J. Biol. Sci. 29, 3822–3829 (2022).
Yadalam, P. K. et al. Gene Interaction Network Analysis Reveals IFI44L as a drug target in Rheumatoid Arthritis and Periodontitis. Molecules 27, 2749 (2022).
Ghosh, N., Saha, I. & Sharma, N. Interactome of human and SARS-CoV-2 proteins to identify human hub proteins associated with comorbidities. Comput. Biol. Med. 138, 104889 (2021).
Kolur, V., Vastrad, B., Vastrad, C., Kotturshetti, S. & Tengli, A. Identification of candidate biomarkers and therapeutic agents for heart failure by bioinformatics analysis. BMC Cardiovasc. Disord. 21, 329 (2021).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Li, J. et al. Machine learning predicts portal vein thrombosis after splenectomy in patients with portal hypertension: comparative analysis of three practical models. World J. Gastroenterol. 28, 4681–4697 (2022).
Gopalakrishnan, D., Sarode, S. C., Sarode, G. S. & Sengupta, N. COVID-19 and oral cancer: critical viewpoint. World J. Clin. Oncol. 13, 725–728 (2022).
Prabani, K. I. P., Weerasekara, I. & Damayanthi, H. D. W. T. COVID-19 vaccine acceptance and hesitancy among patients with cancer: a systematic review and meta-analysis. Public. Health. 212, 66–75 (2022).
Cao, B. et al. Oral simnotrelvir for adult patients with mild-to-moderate Covid-19. N Engl. J. Med. 390, 230–241 (2024).
Huang, N. et al. SARS-CoV-2 infection of the oral cavity and saliva. Nat. Med. 27, 892–903 (2021).
Drożdżal, S. et al. An update on drugs with therapeutic potential for SARS-CoV-2 (COVID-19) treatment. Drug Resist. Updat. 59, 100794 (2021).
Gupta, V. et al. Expression of aldehyde dehydrogenase 1A1 in oral squamous cell carcinoma and its correlation with clinicopathological parameters. Nat. J. Maxillofac. Surg. 13, 208–215 (2022).
Szafarowski, T. et al. Assessment of cancer stem cell marker expression in primary head and neck squamous cell carcinoma shows prognostic value for aldehyde dehydrogenase (ALDH1A1). Eur. J. Pharmacol. 867, 172837 (2020).
Kogan, E. A. et al. ALDH1, CD133, CD34-positive cancer stem cells in lung adenocarcinoma in patients who had a new coronavirus infection and retained the persistence of viral proteins in the lung tissue. Arkh. Patol. 86, 5–14 (2024).
Xue, Q. et al. Copper metabolism in cell death and autophagy. Autophagy 19, 2175–2195 (2023).
Gao, L. et al. Identification of the susceptibility genes for COVID-19 in lung adenocarcinoma with global data and biological computation methods. Comput. Struct. Biotechnol. J. 19, 6229–6239 (2021).
Yadalam, P. K. et al. Machine learning predicts patient tangible outcomes after Dental Implant surgery. IEEE Access. ;10. (2022).
Kumar, V. S. et al. Machine learning in the detection of Dental Cyst, Tumor, and Abscess Lesions. BMC Oral Health. 23, 833 (2023).
Qureshi, R. et al. Machine learning based personalized drug response prediction for lung cancer patients. Sci. Rep. 12, 18935 (2022).
Ngiam, K. Y. & Khor, I. W. Big data and machine learning algorithms for health-care delivery. Lancet Oncol. 20, e262–e273 (2019).
Xu, L., Ru, X. & Song, R. Application of machine learning for drug–target Interaction Prediction. Front. Genet. 12, 680117 (2021).
Hudson, I. L. Data integration using advances in machine learning in Drug Discovery and Molecular Biology. Methods Mol. Biol. 2190, 167–184 (2021).
Koo, C. L., Liew, M. J., Mohamad, M. S. & Salleh, A. H. M. A review for detecting gene-gene interactions using machine learning methods in genetic epidemiology. Biomed. Res. Int. 2013, 432375 (2013).
McKinney, B. A., Reif, D. M., Ritchie, M. D. & Moore, J. H. Machine learning for detecting gene-gene interactions: a review. Appl. Bioinf. 5, 77–88 (2006).
Acknowledgements
We would like to thank the Center of Medical and Bioallied Health Sciences and Research, Ajman University, Ajman, UAE.
Author information
Authors and Affiliations
Contributions
Conceptualization, PY, DA, PN and CA; Data curation, PY, DA, PN and CA; Formal analysis, PY, DA, PN and CA; Funding acquisition, PN; Investigation, PY, DA, PN and CA; Methodology, PY, DA, PN and CA; Project administration, PY; Resources, PY and PN; Software, PY; Supervision, PY, DA, PN and CA; Validation, PY, DA, PN and CA; Visualization, PY, DA, PN and CA; Writing – original draft, PY, DA, PN and CA; Writing – review & editing, PY, DA, PN and CA. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yadalam, P.K., Arumuganainar, D., Natarajan, P.M. et al. Predicting the hub interactome of COVID-19 and oral squamous cell carcinoma: uncovering ALDH-mediated Wnt/β-catenin pathway activation via salivary inflammatory proteins. Sci Rep 15, 4068 (2025). https://doi.org/10.1038/s41598-025-88819-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-88819-2
