

Two Ligand-Binding Sites on SARS-CoV-2 Non-Structural Protein 1 Revealed by Fragment-Based X-ray Screening
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
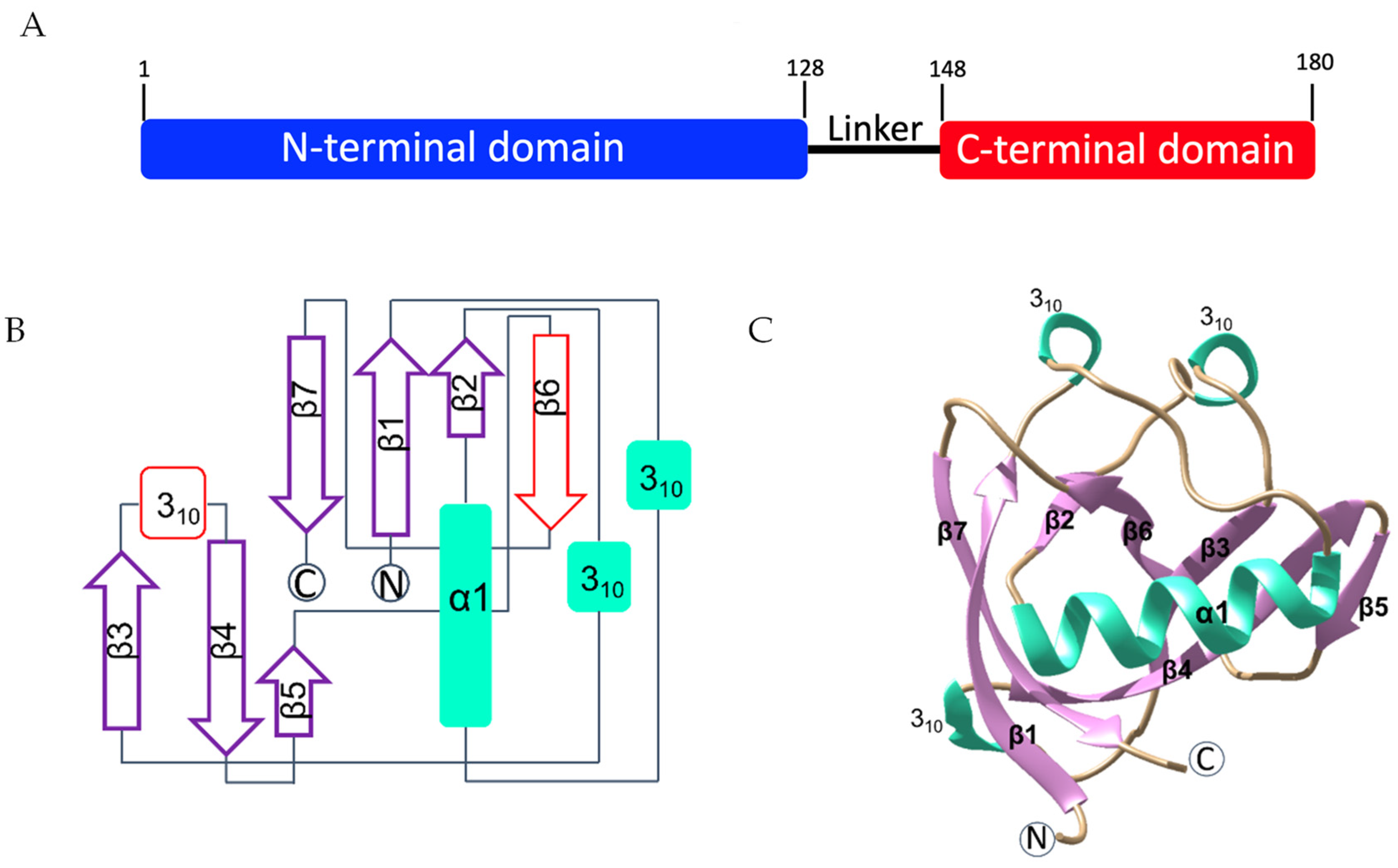
2.1. Structure Determination of Nsp1 to Atomic Resolution
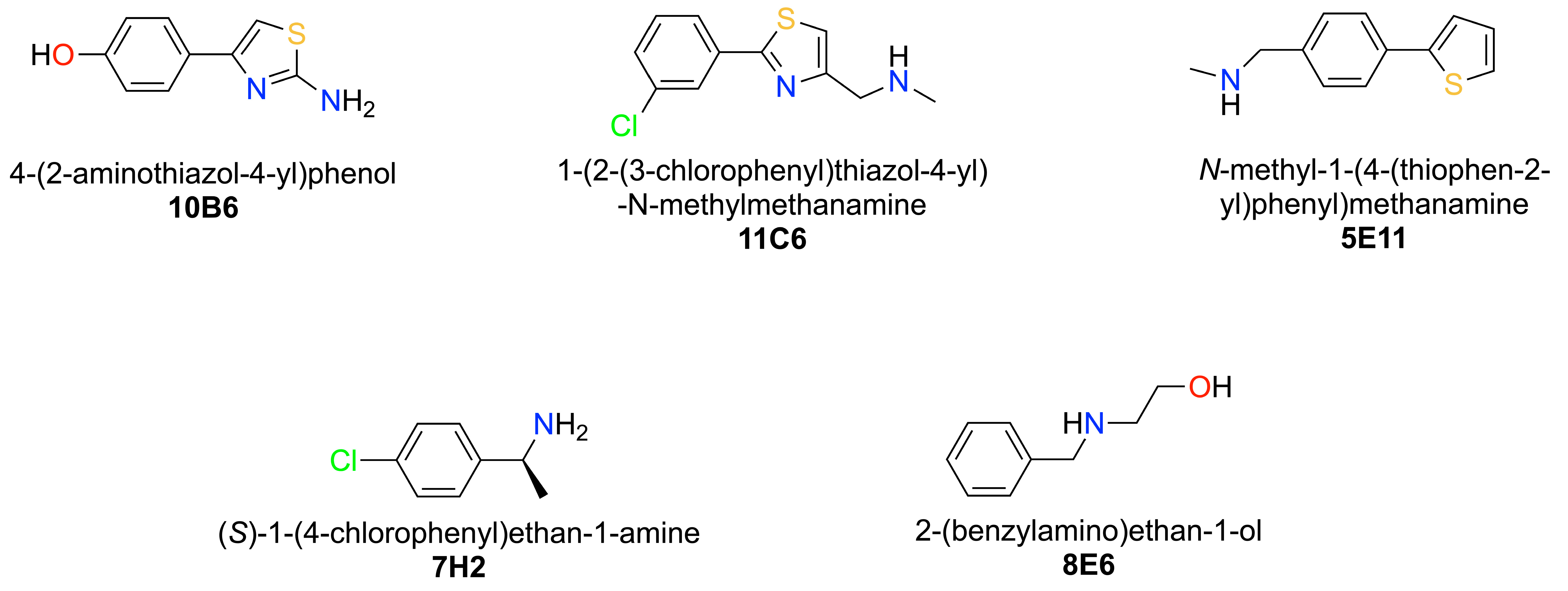
2.2. Fragment Screening via X-ray Crystallography
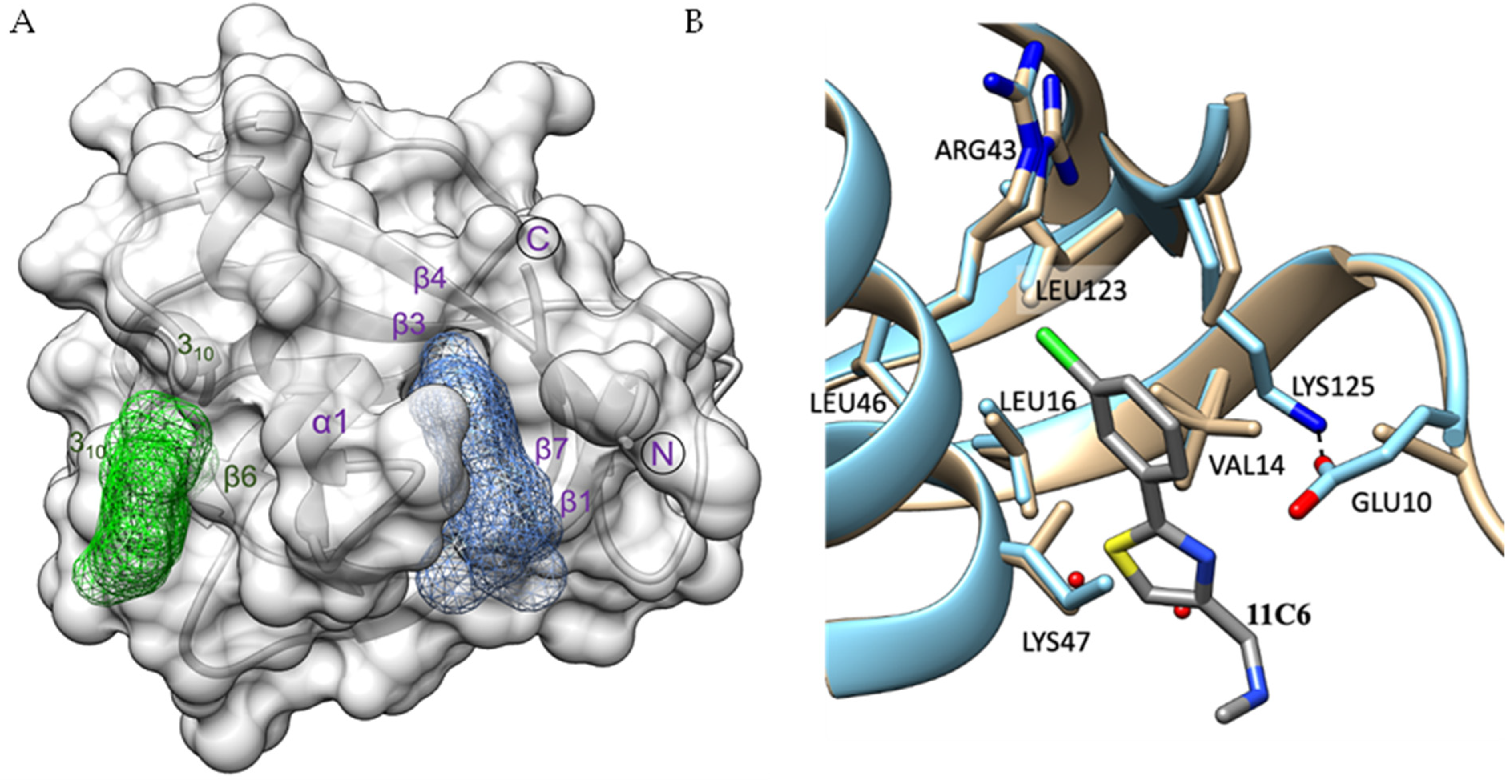
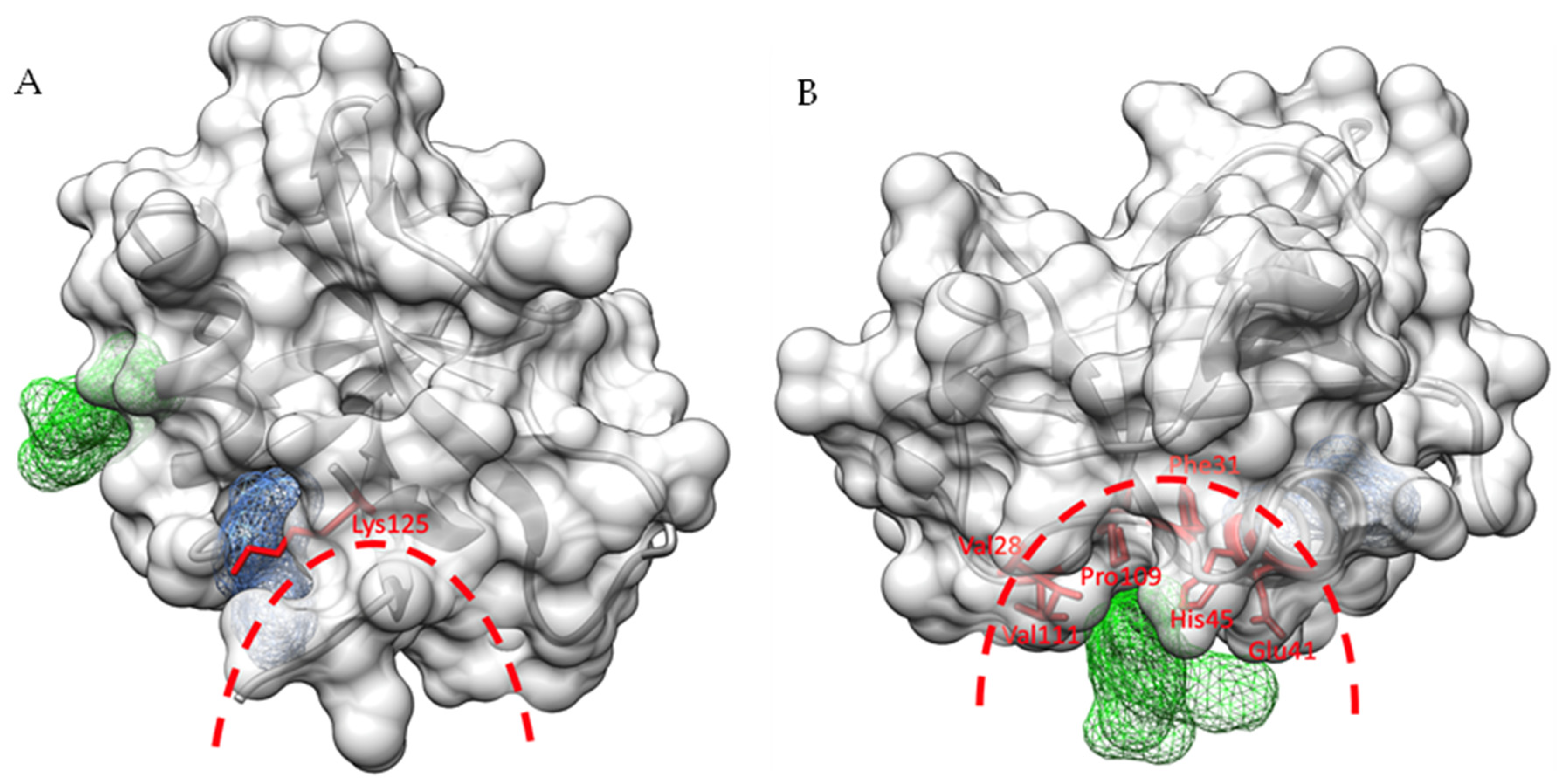
2.3. Identification of Ligand Binding Sites in SARS-CoV-2 Nsp1
2.4. Fragment Hits Binding to Pocket I Induce Small Structural Rearrangements
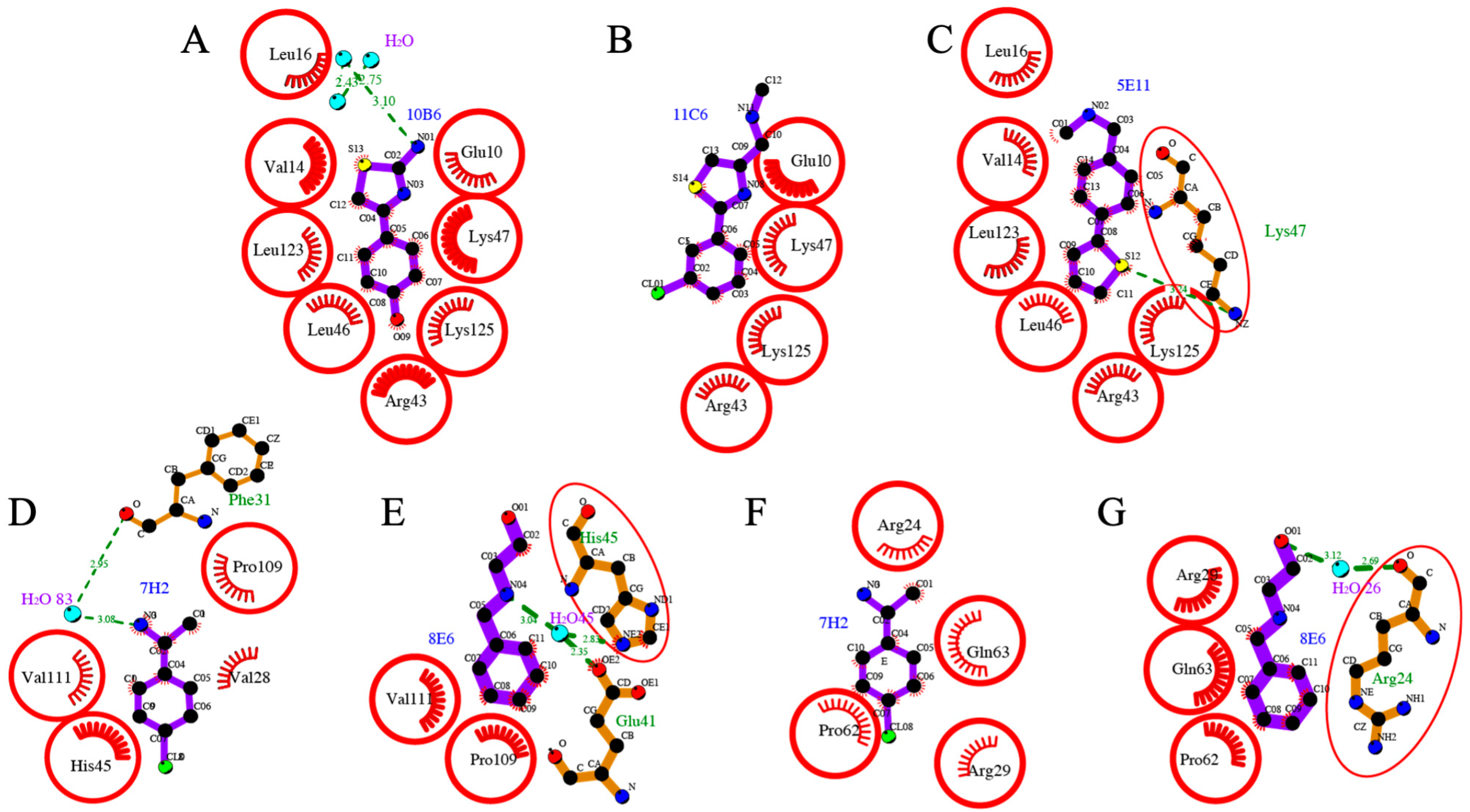
2.5. Detailed Description of SARS-CoV-2 Nsp110-126-Fragment Interactions
2.6. Orthogonal Biophysical Assays to Further Validate Fragment Hits
2.7. Cross Binding of SARS-CoV-2 nsp110-126 Fragment Hits on SARS-CoV-1 and MERS Homologues
2.8. The Two Novel Binding Sites in SARS-CoV-2 Nsp110-126 Are Not Conserved among SARS-CoV-1 Nsp1 and MERS Nsp1
2.9. The Predicted RNA Binding and Validated DNA Polymerase α–Primase Binding Regions Are in Close Proximity to Fragment Binding Sites I and II
2.10. Development of Resistance through Mutations in Nsp1 Ligand Binding Pockets?
2.11. The Rationale for Targeting N- and C-Terminal Domains of Nsp1
3. Methods and Materials
3.1. Subcloning, Expression and Purification of SARS-CoV-2 Nsp1 Constructs
3.2. Crystallisation of Nsp1
3.3. Fragment Soaking of Nsp1 Crystals
3.4. X-ray Diffraction Data Collection, Structure Determination and Refinement
3.5. Thermal Shift Assays for Nsp1-Fragment Complexes Using nanoDSF
3.6. MST for the Estimation of Kd Values of Fragment Hits
3.7. Calculation of Physico-Chemical Properties and Preparation of Figures
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zhong, N.S.; Zheng, B.J.; Li, Y.M.; Poon, L.L.M.; Xie, Z.H.; Chan, K.H.; Li, P.H.; Tan, S.Y.; Chang, Q.; Xie, J.P.; et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Newman, C.; Buesching, C.D.; Macdonald, D.W.; Zhou, Z.M. Animal sales from Wuhan wet markets immediately prior to the COVID-19 pandemic. Sci. Rep. 2021, 11, 11898. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Zhang, S.F.; Li, W.; Zhang, L.; Cheung, T.; Tang, Y.L.; Ng, C.H.; Yang, B.X.; Xiang, Y.T. Mental health status and quality of life in close contacts of COVID-19 patients in the post-COVID-19 era: A comparative study. Transl. Psychiatry 2021, 11, 505. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Yamana, T.K.; Kandula, S.; Galanti, M.; Shaman, J. Burden and characteristics of COVID-19 in the United States during 2020. Nature 2021, 598, 338–342. [Google Scholar] [CrossRef]
- Josephson, A.; Kilic, T.; Michler, J.D. Socioeconomic impacts of COVID-19 in low-income countries. Nat. Hum. Behav. 2021, 5, 557–565. [Google Scholar] [CrossRef]
- Salje, H.; Kiem, C.T.; Lefrancq, N.; Courtejoie, N.; Bosetti, P.; Paireau, J.; Andronico, A.; Hozé, N.; Richet, J.; Dubost, C.-L.; et al. Estimating the burden of SARS-CoV-2 in France. Science 2020, 369, 208–211. [Google Scholar] [CrossRef]
- Aruffo, E.; Yuan, P.; Tan, Y.; Gatov, E.; Gournis, E.; Collier, S.; Ogden, N.; Bélair, J.; Zhu, H. Community structured model for vaccine strategies to control COVID-19 spread: A mathematical study. medRxiv 2021. [Google Scholar] [CrossRef]
- Porter, J.R. Vaccines, social measures and COVID-19—A European evidence-based analysis Vaccines, social measures and COVID-19. medRxiv 2021. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Lavine, C.L.; Rawson, S.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; et al. Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science 2021, 373, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Abu-Raddad, L.J.; Chemaitelly, H.; Butt, A.A. Effectiveness of the BNT162b2 COVID-19 Vaccine against the B. 1.1. 7 and B. 1.351 Variants. N. Engl. J. Med. 2021, 385, 187–189. [Google Scholar] [CrossRef]
- Lipsitch, M.; Dean, N.E. Understanding COVID-19 vaccine efficacy. Science 2020, 370, 763–765. [Google Scholar] [CrossRef]
- Madison, A.A.; Shrout, M.R.; Renna, M.E.; Kiecolt-Glaser, J.K. Psychological and behavioral predictors of vaccine efficacy: Considerations for COVID-19. Perspect. Psychol. Sci. 2021, 16, 191–203. [Google Scholar] [CrossRef]
- The Pfizer BioNTech (BNT162b2) COVID-19 vaccine: What You Need to Know. Available online: https://www.who.int/news-room/feature-stories/detail/who-can-take-the-pfizer-biontech-covid-19--vaccine?adgroupsurvey={adgroupsurvey}&gclid=EAIaIQobChMIqpiuz4u08wIVkOmyCh0UlQ2KEAAYASABEgIIxvD_BwE (accessed on 2 September 2021).
- Taha, H.R.; Keewan, N.; Slati, F.; Al-Sawalha, N. Remdesivir: A Closer Look at Its Effect in COVID-19 Pandemic. Pharmacology 2021, 106, 462–468. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Treatment Guidelines Panel. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. National Institutes of Health. Available online: https://www.covid19treatmentguidelines.nih.gov/ (accessed on 27 September 2022).
- Costanzo, M.; de Giglio, M.A.R.; Roviello, G.N. SARS-CoV-2: Recent Reports on Antiviral Therapies Based on Lopinavir/Ritonavir, Darunavir/Umifenovir, Hydroxychloroquine, Remdesivir, Favipiravir and other Drugs for the Treatment of the New Coronavirus. Curr. Med. Chem. 2020, 27, 4536–4541. [Google Scholar] [CrossRef]
- Moreno, S.; Alcázar, B.; Dueñas, C.; del Castillo, J.G.; Antela, J.O. Use of Antivirals in SARS-CoV-2 Infection. Critical Review of the Role of Remdesivir. Drug Des. Dev. Ther. 2022, 16, 827–841. [Google Scholar]
- Parvathaneni, V.; Gupta, V. Utilizing drug repurposing against COVID-19—Efficacy, limitations, and challenges. Life Sci. 2020, 259, 118275. [Google Scholar] [CrossRef]
- Vegivinti, C.T.R.; Evanson, K.W.; Lyons, H.; Akosman, I.; Barrett, A.; Hardy, N.; Kane, B.; Keesari, P.R.; Pulakurthi, Y.S.; Sheffels, E.; et al. Efficacy of antiviral therapies for COVID-19: A systematic review of randomized controlled trials. BMC Infect. Dis. 2022, 22, 107. [Google Scholar] [CrossRef]
- Kumar, D.; Trivedi, N. Disease-drug and drug-drug interaction in COVID-19: Risk and assessment. Biomed. Pharmacother. 2021, 139, 111642. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, R.; Ashraf, R.; Rowe, H.; Zipursky, J.; Clarfield, L.; Maxwell, C.; Arzola, C.; Lapinsky, S.; Paquette, K.; Murthy, S.; et al. Pregnancy and COVID-19: Pharmacologic considerations. Ultrasound Obstet. Gynecol. 2021, 57, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [PubMed]
- Min, Y.Q.; Mo, Q.; Wang, J.; Deng, F.; Wang, H.; Ning, Y.J. SARS-CoV-2 nsp1: Bioinformatics, Potential Structural and Functional Features, and Implications for Drug/Vaccine Designs. Front. Microbiol. 2020, 11, 587317. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Huang, C.; Lokugamage, K.; Kamitani, W.; Ikegami, T.; Tseng, C.T.; Makino, S. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 2008, 82, 4471–4479. [Google Scholar] [CrossRef] [Green Version]
- Terada, Y.; Kawachi, K.; Matsuura, Y.; Kamitani, W. MERS coronavirus nsp1 participates in an efficient propagation through a specific interaction with viral RNA. Virology 2017, 511, 95–105. [Google Scholar] [CrossRef]
- Lapointe, C.P.; Grosely, R.; Johnson, A.G.; Wang, J.; Fernandez, I.S.; Puglisi, J.D. Dynamic competition between SARS-CoV-2 NSP1 and mRNA on the human ribosome inhibits translation initiation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017715118. [Google Scholar] [CrossRef]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.A.; Leibundgut, M.; Thiel, V.; Muhlemann, O.; Ban, N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Shi, M.; Wang, L.; Fontana, P.; Vora, S.; Zhang, Y.; Fu, T.M.; Lieberman, J.; Wu, H. SARS-CoV-2 Nsp1 suppresses host but not viral translation through a bipartite mechanism. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci. Adv. 2021, 7, eabe7386. [Google Scholar] [CrossRef]
- Lokugamage, K.G.; Narayanan, K.; Nakagawa, K.; Terasaki, K.; Ramirez, S.I.; Tseng, C.T.; Makino, S. Middle East Respiratory Syndrome Coronavirus nsp1 Inhibits Host Gene Expression by Selectively Targeting mRNAs Transcribed in the Nucleus while Sparing mRNAs of Cytoplasmic Origin. J. Virol. 2015, 89, 10970–10981. [Google Scholar] [CrossRef] [PubMed]
- Semper, C.; Watanabe, N.; Savchenko, A. Structural characterization of nonstructural protein 1 from SARS-CoV-2. iScience 2021, 24, 101903. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.K.; Green, T.J.; Petit, C.M. Structure of Nonstructural Protein 1 from SARS-CoV-2. J. Virol. 2021, 95, e02019-20. [Google Scholar] [CrossRef] [PubMed]
- Svensson, O.; Gilski, M.; Nurizzo, D.; Bowler, M.W. Multi-position data collection and dynamic beam sizing: Recent improvements to the automatic data-collection algorithms on MASSIF-1. Acta Crystallogr. Sect. D 2018, 74, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Bowler, M.W.; Nurizzo, D.; Barrett, R.; Beteva, A.; Bodin, M.; Caserotto, H.; Delageniere, S.; Dobias, F.; Flot, D.; Giraud, T.; et al. MASSIF-1: A beamline dedicated to the fully automatic characterization and data collection from crystals of biological macromolecules. J. Synchrotron Radiat. 2015, 22, 1540–1547. [Google Scholar] [CrossRef]
- Pearce, N.M.; Krojer, T.; Bradley, A.R.; Collins, P.; Nowak, R.P.; Talon, R.; Marsden, B.D.; Kelm, S.; Shi, J.; Deane, C.M.; et al. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 2017, 8, 15123. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Kozielski, F.; Sele, C.; Talibov, V.O.; Lou, J.; Dong, D.; Wang, Q.; Shi, X.; Nyblom, M.; Rogstam, A.; Krojer, T.; et al. Identification of fragments binding to SARS-CoV-2 nsp10 reveals ligand-binding sites in conserved interfaces between nsp10 and nsp14/nsp16. RSC Chem. Biol. 2022, 3, 44–55. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vankadari, N.; Jeyasankar, N.N.; Lopes, W.J. Structure of the SARS-CoV-2 Nsp1/5′-Untranslated Region Complex and Implications for Potential Therapeutic Targets, a Vaccine, and Virulence. J. Phys. Chem. Lett. 2020, 11, 9659–9668. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L. The Pol α-Primase Complex. In The Eukaryotic Replisome: A Guide to Protein Structure and Function; MacNeill, S., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 157–169. [Google Scholar]
- Starokadomskyy, P.; Escala Perez-Reyes, A.; Burstein, E. Immune Dysfunction in Mendelian Disorders of POLA1 Deficiency. J. Clin. Immunol. 2021, 41, 285–293. [Google Scholar] [CrossRef]
- Starokadomskyy, P.; Gemelli, T.; Rios, J.J.; Xing, C.; Wang, R.C.; Li, H.; Pokatayev, V.; Dozmorov, I.; Khan, S.; Miyata, N.; et al. DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat. Immunol. 2016, 17, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilkenny, M.L.; Veale, C.E.; Guppy, A.; Hardwick, S.W.; Chirgadze, D.Y.; Rzechorzek, N.J.; Maman, J.D.; Pellegrini, L. Structural basis for the interaction of SARS-CoV-2 virulence factor nsp1 with DNA polymerase alpha-primase. Protein Sci. 2022, 31, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Mou, K.; Mukhtar, F.; Khan, M.T.; Darwish, D.B.; Peng, S.; Muhammad, S.; Al-Sehemi, A.G.; Wei, D.-Q. Emerging Mutations in Nsp1 of SARS-CoV-2 and Their Effect on the Structural Stability. Pathogens 2021, 10, 1285. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, A.; Kumar, P.; Garg, N.; Giri, R. SARS-CoV-2 NSP1 C-terminal (residues 131–180) is an intrinsically disordered region in isolation. Curr. Res. Virol. Sci. 2021, 2, 100007. [Google Scholar] [CrossRef]
- Mendez, A.S.; Ly, M.; Gonzalez-Sanchez, A.M.; Hartenian, E.; Ingolia, N.T.; Cate, J.H.; Glaunsinger, B.A. The N-terminal domain of SARS-CoV-2 nsp1 plays key roles in suppression of cellular gene expression and preservation of viral gene expression. Cell Rep. 2021, 37, 109841. [Google Scholar] [CrossRef]
- Kamitani, W.; Narayanan, K.; Huang, C.; Lokugamage, K.; Ikegami, T.; Ito, N.; Kubo, H.; Makino, S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 12885–12890. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, W.; Huang, C.; Narayanan, K.; Lokugamage, K.G.; Makino, S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009, 16, 1134–1140. [Google Scholar] [CrossRef]
- Huang, C.; Lokugamage, K.G.; Rozovics, J.M.; Narayanan, K.; Semler, B.L.; Makino, S. SARS Coronavirus nsp1 Protein Induces Template-Dependent Endonucleolytic Cleavage of mRNAs: Viral mRNAs Are Resistant to nsp1-Induced RNA Cleavage. PLoS Pathog. 2011, 7, e1002433. [Google Scholar] [CrossRef] [Green Version]
- Shuvalov, A.; Shuvalova, E.; Biziaev, N.; Sokolova, E.; Evmenov, K.; Pustogarov, N.; Arnautova, A.; Matrosova, V.; Egorova, T.; Alkalaeva, E. Nsp1 of SARS-CoV-2 stimulates host translation termination. RNA Biol. 2021, 18, 804–817. [Google Scholar] [CrossRef]
- Borsatto, A.; Akkad, O.; Galdadas, I.; Ma, S.; Damfo, S.; Haider, S.; Kozielski, F.; Estarellas, C.; Gervasio, F.L. Revealing druggable cryptic pockets in the Nsp-1 of SARS-CoV-2 and other β-coronaviruses by simulations and crystallography. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D 2011, 67, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonrhein, C.; Tickle, I.J.; Flensburg, C.; Keller, P.; Paciorek, W.; Sharff, A.; Bricogne, G. Advances in automated data analysis and processing within autoPROC, combined with improved characterisation, mitigation and visualisation of the anisotropy of diffraction limits using STARANISO. Acta Crystallogr. Sect. A 2018, 74, a360. [Google Scholar] [CrossRef] [Green Version]
- Krug, M.; Weiss, M.S.; Heinemann, U.; Mueller, U. XDSAPP: A graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 2012, 45, 568–572. [Google Scholar] [CrossRef]
- Winter, G.; Beilsten-Edmands, J.; Devenish, N.; Gerstel, M.; Gildea, R.J.; McDonagh, D.; Pascal, E.; Waterman, D.G.; Williams, B.H.; Evans, G. DIALS as a toolkit. Protein Sci. 2022, 31, 232–250. [Google Scholar] [CrossRef]
- Incardona, M.-F.; Bourenkov, G.P.; Levik, K.; Pieritz, R.A.; Popov, A.N.; Svensson, O. EDNA: A framework for plugin-based applications applied to X-ray experiment online data analysis. J. Synchrotron Radiat. 2009, 16, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallographica. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection and Refinement Statistics | SARS-CoV-2 nsp110-126 |
|---|---|
| PDB entry | 8A55 |
| Wavelength [Å] | 0.82656 |
| Resolution range [Å] | 32.63–0.99 (1.03–0.99) |
| Space group | P43212 |
| Unit cell parameters [Å, °] | 36.81, 36.81, 140.96, 90, 90, 90 |
| Molecules per asymmetric unit | 1 |
| Total reflections | 421599 (39143) |
| Unique reflections | 55025 (5335) |
| Multiplicity | 7.7 (7.3) |
| Completeness [%] | 99.5 (98.4) |
| Mean I/sigma(I) | 12.7 (0.8) |
| Wilson B-factor (Å2) | 13.5 |
| Rmeas [%] | 6.2 (206.9) |
| CC1/2 | 0.998 (0.267) |
| Reflections used in refinement | 55001 (5317) |
| Rcryst/Rfree [%] | 16.3 (32.2)/18.1 (33.4) |
| Total no. of non-hydrogen atoms (protein) | 1108 |
| No. of protein/solvent residues | 976/132 |
| RMSD bonds length, bond angles [Å, °] | 0.011/1.2 |
| Ramachandran favoured/allowed/outliers/rotamer outliers [%] | 98.38/1.7/0.0/0.9 |
| Clashscore | 7.5 |
| Average B-factor/protein/solvent | 23.6/21.9/36.3 |
| Binding Site | Fragments | Residues |
|---|---|---|
| I | 10B6, 11C6, 5E11 | Glu10, Val14, Leu16, Arg43, Leu46, Lys47, Leu123 and Lys125 |
| II | 7H2, 8E6 | Val28, Phe31, Glu41, His45, Pro109, Val111; Symmetry mate: Arg24, Arg29, Pro62, Gln63 |
| Fragment ID | Binding Site | MST Kd [mM] | TSA Ti *± SD | TSA ΔTi [°C] | TSA |ΔTi|-3SD [°C] | MW [Da] | MolLogP (mol) | HBA & HBD [No] | mol PSA [Å2] | MolLogS [Log(moles/L)] |
|---|---|---|---|---|---|---|---|---|---|---|
| 5E11 | I | 0.48 ± 0.30 | APUC | / | / | 203.1 | 2.75 | 1, 1 | 13.0 | −3.526 |
| 10B6 | I | 0.56 ± 0.19 | 51.27 ± 0.15 | −2.79 | 2.33 | 192.0 | 2.48 | 2, 3 | 48.4 | −3.495 |
| 11C6 | I | 1.16 ± 0.22 | 53.78 ± 0.05 | −0.27 | 0.13 | 238.0 | 2.73 | 2, 1 | 22.4 | −4.518 |
| 7H2 | II | >20 | 53.55 ± 0.25 | −0.50 | −0.25 | 155.1 | 1.23 | 1, 2 | 20.6 | −2.565 |
| 8E6 | II | 8.32 ± 3.32 | 52.72 ± 0.22 | −1.33 | 0.67 | 151.1 | 0.51 | 2, 2 | 28.8 | −0.603 |
| Fragment Hit | SARS-CoV-2 nsp1 MST, Kd [mM] | SARS-CoV-1 nsp1 MST, Kd [mM] | MERS nsp1 MST, Kd [mM] |
|---|---|---|---|
| 5E11 | 0.48 ± 0.30 | 12.76 ± 24.13 | >20 |
| 10B6 | 0.56 ± 0.19 | >20 | 4.92 ± 4.19 |
| 11C6 | 1.16 ± 0.22 | 0.38 ± 0.14 | n.i. |
| 7H2 | >20 | n.i. | n.i |
| 8E6 | 8.32 ± 3.32 | >20 | >20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Damfo, S.; Lou, J.; Pinotsis, N.; Bowler, M.W.; Haider, S.; Kozielski, F. Two Ligand-Binding Sites on SARS-CoV-2 Non-Structural Protein 1 Revealed by Fragment-Based X-ray Screening. Int. J. Mol. Sci. 2022, 23, 12448. https://doi.org/10.3390/ijms232012448
Ma S, Damfo S, Lou J, Pinotsis N, Bowler MW, Haider S, Kozielski F. Two Ligand-Binding Sites on SARS-CoV-2 Non-Structural Protein 1 Revealed by Fragment-Based X-ray Screening. International Journal of Molecular Sciences. 2022; 23(20):12448. https://doi.org/10.3390/ijms232012448
Chicago/Turabian StyleMa, Shumeng, Shymaa Damfo, Jiaqi Lou, Nikos Pinotsis, Matthew W. Bowler, Shozeb Haider, and Frank Kozielski. 2022. "Two Ligand-Binding Sites on SARS-CoV-2 Non-Structural Protein 1 Revealed by Fragment-Based X-ray Screening" International Journal of Molecular Sciences 23, no. 20: 12448. https://doi.org/10.3390/ijms232012448