
Computational Analysis of SAM Analogs as Methyltransferase Inhibitors of nsp16/nsp10 Complex from SARS-CoV-2
 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Structural Analysis of MD Simulations and PCA Analysis
2.2. Binding Free Energy and Residual Decomposition Analysis
3. Computational Methods
3.1. System Setup and MD Simulations
3.2. Structural and Thermodynamic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- WHO. COVID-19 Weekly Epidemiological Update. 2020. Available online: https://covid19.who.int/ (accessed on 30 October 2022).
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. Chapter Three—The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. In Coronaviruses; Ziebuhr, J., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 96, pp. 59–126. ISBN 0065-3527. [Google Scholar]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. In Vitro Reconstitution of SARS-Coronavirus mRNA Cap Methylation. PLoS Pathog. 2010, 6, e1000863. [Google Scholar] [CrossRef]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat. Commun. 2020, 11, 3717. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.R.A.; Urban, J.; Araújo, E.; Lameira, J.; Moliner, V.; Alves, C.N. Exploring the Catalytic Mechanism of the RNA Cap Modification by nsp16-nsp10 Complex of SARS-CoV-2 through a QM/MM Approach. Int. J. Mol. Sci. 2021, 23, 300. [Google Scholar] [CrossRef]
- Anjum, F.; Mohammad, T.; Asrani, P.; Shafie, A.; Singh, S.; Yadav, D.K.; Uversky, V.N.; Hassan, M.I. Identification of intrinsically disorder regions in non-structural proteins of SARS-CoV-2: New insights into drug and vaccine resistance. Mol. Cell. Biochem. 2022, 477, 1607–1619. [Google Scholar] [CrossRef]
- Bobiļeva, O.; Bobrovs, R.; Kaņepe, I.; Patetko, L.; Kalniņš, G.; Šišovs, M.; Bula, A.L.; Gri̅nberga, S.; Borodušķis, M.; Ramata-Stunda, A.; et al. Potent SARS-CoV-2 mRNA Cap Methyltransferase Inhibitors by Bioisosteric Replacement of Methionine in SAM Cosubstrate. ACS Med. Chem. Lett. 2021, 12, 1102–1107. [Google Scholar] [CrossRef]
- Chen, Y.; Su, C.; Ke, M.; Jin, X.; Xu, L.; Zhang, Z.; Wu, A.; Sun, Y.; Tien, P.; Ahola, T.; et al. Biochemical and Structural Insights into the Mechanisms of SARS Coronavirus RNA Ribose 2′-O-Methylation by nsp16/nsp10 Protein Complex. PLoS Pathog. 2011, 7, e1002294. [Google Scholar] [CrossRef]
- Decroly, E.; Imbert, I.; Coutard, B.; Bouvet, M.; Selisko, B.; Alvarez, K.; Gorbalenya, A.E.; Snijder, E.J.; Canard, B. Coronavirus Nonstructural Protein 16 Is a Cap-0 Binding Enzyme Possessing (Nucleoside-2′O)-Methyltransferase Activity. J. Virol. 2008, 82, 8071–8084. [Google Scholar] [CrossRef] [Green Version]
- Aouadi, W.; Blanjoie, A.; Vasseur, J.-J.; Debart, F.; Canard, B.; Decroly, E. Binding of the Methyl Donor S-Adenosyl-l-Methionine to Middle East Respiratory Syndrome Coronavirus 2′-O-Methyltransferase nsp16 Promotes Recruitment of the Allosteric Activator nsp10. J. Virol. 2017, 91, 2217–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, J.L.; Diamond, M.S. Innate immune restriction and antagonism of viral RNA lacking 2′-O methylation. Virology 2015, 479–480, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, T.; Arya, S.; Chan, S.-H.; Qi, S.; Dai, N.; Misra, A.; Park, J.-G.; Oladunni, F.; Kovalskyy, D.; Hromas, R.A.; et al. Structural basis of RNA cap modification by SARS-CoV-2. Nat. Commun. 2020, 11, 3718. [Google Scholar] [CrossRef] [PubMed]
- Araújo, E.; Lima, A.H.; Lameira, J. Catalysis by solvation rather than the desolvation effect: Exploring the catalytic efficiency of SAM-dependent chlorinase. Phys. Chem. Chem. Phys. 2017, 19, 21350–21356. [Google Scholar] [CrossRef] [PubMed]
- Lameira, J.; Bora, R.P.; Chu, Z.T.; Warshel, A. Methyltransferases do not work by compression, cratic, or desolvation effects, but by electrostatic preorganization. Proteins Struct. Funct. Bioinform. 2015, 83, 218–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, N.; Lameira, J.; Roge, J.; Lima, A.H. Exploring Chloride Selectivity and Halogenase Regioselectivity of the SalL Enzyme through Quantum Mechanical/Molecular Mechanical Modeling. J. Chem. Inf. Model. 2020, 60, 738–746. [Google Scholar] [CrossRef]
- Roca, M.; Andrés, J.; Moliner, V.; Tuñón, I.; Bertrán, J. On the Nature of the Transition State in Catechol O-Methyltransferase. A Complementary Study Based on Molecular Dynamics and Potential Energy Surface Explorations. J. Am. Chem. Soc. 2005, 127, 10648–10655. [Google Scholar] [CrossRef] [PubMed]
- Świderek, K.; Tuñón, I.; Williams, I.H.; Moliner, V. Insights on the Origin of Catalysis on Glycine N-Methyltransferase from Computational Modeling. J. Am. Chem. Soc. 2018, 140, 4327–4334. [Google Scholar] [CrossRef]
- Sulimov, A.; Kutov, D.; Ilin, I.; Xiao, Y.; Jiang, S.; Sulimov, V. Novel Inhibitors of 2′-O-Methyltransferase of the SARS-CoV-2 Coronavirus. Molecules 2022, 27, 2721. [Google Scholar] [CrossRef]
- Eissa, I.H.; Alesawy, M.S.; Saleh, A.M.; Elkaeed, E.B.; Alsfouk, B.A.; El-Attar, A.-A.M.M.; Metwaly, A.M. Ligand and Structure-Based In Silico Determination of the Most Promising SARS-CoV-2 nsp16-nsp10 2′-o-Methyltransferase Complex Inhibitors among 3009 FDA Approved Drugs. Molecules 2022, 27, 2287. [Google Scholar] [CrossRef]
- Bobrovs, R.; Kanepe, I.; Narvaiss, N.; Patetko, L.; Kalnins, G.; Sisovs, M.; Bula, A.L.; Grinberga, S.; Boroduskis, M.; Ramata-Stunda, A.; et al. Discovery of SARS-CoV-2 Nsp14 and Nsp16 Methyltransferase Inhibitors by High-Throughput Virtual Screening. Pharmaceuticals 2021, 14, 1243. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate−DNA Helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Sondergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of p K a Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M. Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. Amber18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Sittel, F.; Jain, A.; Stock, G. Principal component analysis of molecular dynamics: On the use of Cartesian vs. internal coordinates. J. Chem. Phys. 2014, 141, 014111. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins. In Protein Dynamics; Livesay, D.R., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 193–226. ISBN 978-1-62703-658-0. [Google Scholar]
- Qayed, W.S.; Ferreira, R.S.; Silva, J.R.A. In Silico Study towards Repositioning of FDA-Approved Drug Candidates for Anticoronaviral Therapy: Molecular Docking, Molecular Dynamics and Binding Free Energy Calculations. Molecules 2022, 27, 5988. [Google Scholar] [CrossRef] [PubMed]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef]
- Srivastava, M.; Mittal, L.; Kumari, A.; Asthana, S. Molecular Dynamics Simulations Reveal the Interaction Fingerprint of Remdesivir Triphosphate Pivotal in Allosteric Regulation of SARS-CoV-2 RdRp. Front. Mol. Biosci. 2021, 8, 639614. [Google Scholar] [CrossRef]
- Sitthiyotha, T.; Chunsrivirot, S. Computational design of SARS-CoV-2 peptide binders with better predicted binding affinities than human ACE2 receptor. Sci. Rep. 2021, 11, 15650. [Google Scholar] [CrossRef]
- Koulgi, S.; Jani, V.; Uppuladinne, M.V.N.; Sonavane, U.; Joshi, R. Remdesivir-bound and ligand-free simulations reveal the probable mechanism of inhibiting the RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2020, 10, 26792–26803. [Google Scholar] [CrossRef]
- Yu, W.; Wu, X.; Zhao, Y.; Chen, C.; Yang, Z.; Zhang, X.; Ren, J.; Wang, Y.; Wu, C.; Li, C.; et al. Computational Simulation of HIV Protease Inhibitors to the Main Protease (Mpro) of SARS-CoV-2: Implications for COVID-19 Drugs Design. Molecules 2021, 26, 7385. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.R.A.; Kruger, H.G.; Molfetta, F.A. Drug repurposing and computational modeling for discovery of inhibitors of the main protease (M pro) of SARS-CoV-2. RSC Adv. 2021, 11, 23450–23458. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
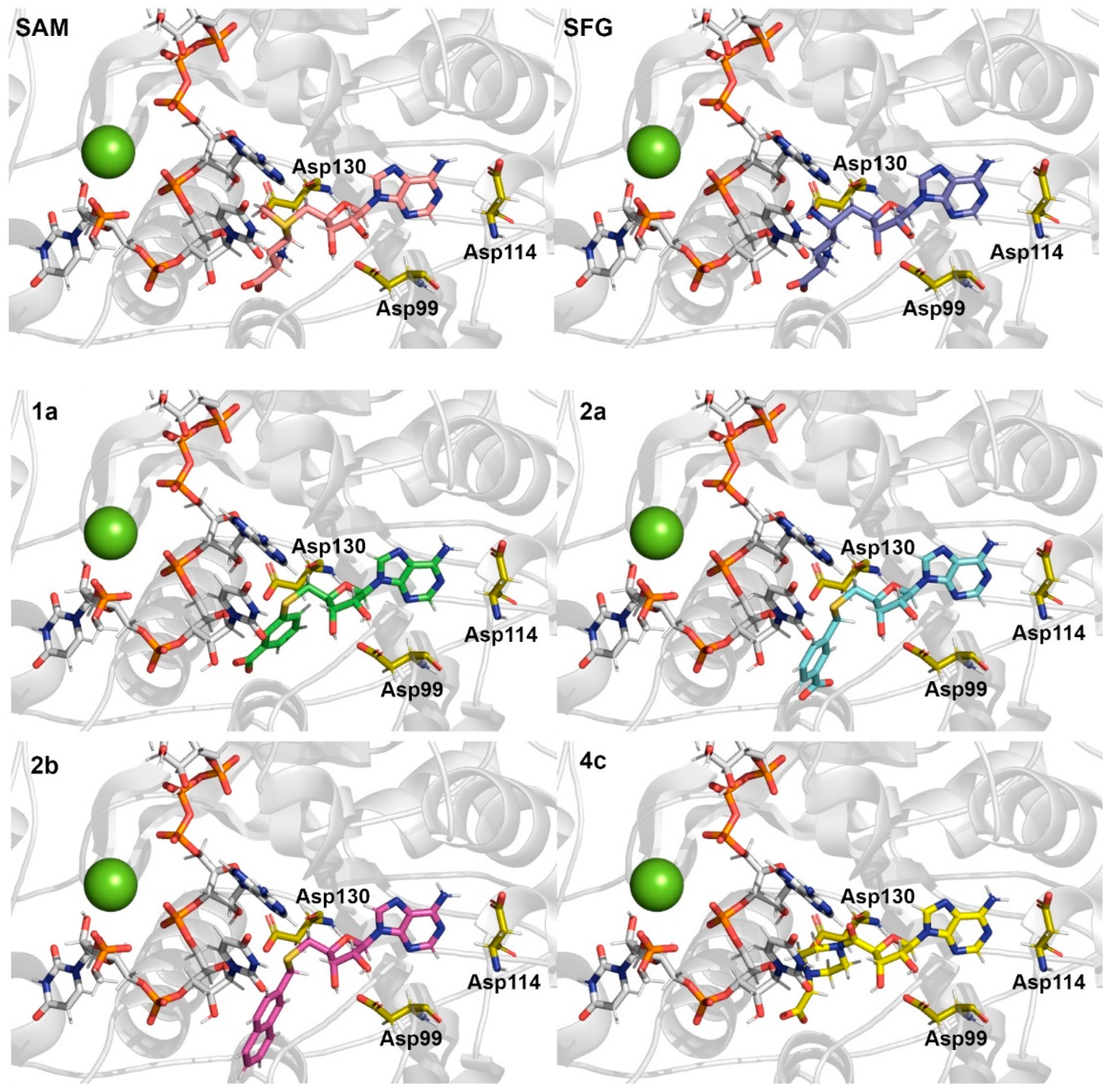
| System | Donor | Acceptor | Average Distance (Å) | Occupancy (%) |
|---|---|---|---|---|
| SAM | SAM(O3) | Asp99(OD1) | 2.68 | 145.51% |
| SAM(N) | Asp130(OD2) | 2.82 | 74.17% | |
| SAM(N6) | Asp114(OD1) | 2.86 | 55.22% | |
| SAM(N) | Gly71(O) | 2.82 | 45.40% | |
| Cys115(N) | SAM(N1) | 2.92 | 15.59% | |
| SFG | SFG(O13) | Asp99(OD1) | 2.71 | 142.09% |
| SFG(N22) | Asp114(OD2) | 2.87 | 43.05% | |
| SFG(N1) | Asp130(OD2) | 2.84 | 25.98% | |
| Cys115(N) | SFG(N16) | 2.91 | 24.18% | |
| 1a | 1a(O3) | Asp99(OD1) | 2.67 | 94.61% |
| Ser74(OG) | 1a(O4) | 2.67 | 70.69% | |
| 1a(N3) | Asp114(OD1) | 2.87 | 65.34% | |
| Asn43(ND2) | 1a(O4) | 2.82 | 21.47% | |
| Asp75(N) | 1a(O5) | 2.88 | 23.41% | |
| Cys115(N) | 1a(N1) | 2.92 | 8.94% | |
| 2a | 2a(O2) | Asp99(OD2) | 2.70 | 91.07% |
| Ser74(OG) | 2a(O01) | 2.66 | 65.94% | |
| 2a(N6) | Asp114(OD2) | 2.86 | 43.06% | |
| Cys115(N) | 2a(N1) | 2.92 | 22.34% | |
| Lys76(NZ) | 2a(O03) | 2.80 | 16.14% | |
| Asn43(ND2) | 2a(O01) | 2.81 | 14.43% | |
| 2b | 2b(O9) | Asp99(OD2) | 2.70 | 82.35% |
| 2b(N22) | Asp114(OD2) | 2.87 | 38.12% | |
| Leu100(N) | 2b(O9) | 2.91 | 19.86% | |
| Cys115(N) | 2b(N12) | 2.92 | 16.75% | |
| Tyr132(N) | 2b(O5) | 2.88 | 4.13% | |
| 4c | 4c(O2) | Asp99(OD1) | 2.65 | 161.55% |
| 4c(N5) | Asp114(OD1) | 2.87 | 41.36% | |
| Lys76(NZ) | 4c(O6) | 2.78 | 12.62% | |
| Cys115(N) | 4c(N2) | 2.92 | 11.74% | |
| Asp75(N) | 4c(O6) | 2.90 | 7.54% | |
| Tyr132(N) | 4c(O5) | 2.88 | 1.67% |
| System | ||||||
|---|---|---|---|---|---|---|
| SAM | −51.2 ± 0.1 | −370.4 ± 0.3 | 358.4 ± 0.2 | −6.8 ± 0.1 | −70.0 ± 0.1 | - |
| SFG | −45.2 ± 0.1 | −83.5 ± 0.3 | 94.8 ± 0.2 | −5.9 ± 0.1 | −39.8 ± 0.1 | −8.3 |
| 1a | −41.8 ± 0.1 | 72.0 ± 0.7 | −71.0 ± 0.5 | −6.0 ± 0.1 | −46.8 ± 0.2 | −10.2 |
| 2a | −48.3 ± 0.1 | 34.8 ± 0.4 | −38.8 ± 0.3 | −6.4 ± 0.1 | −58.7 ± 0.2 | −11.5 |
| 2b | −51.9 ± 0.1 | −54.6 ± 0.3 | 63.2 ± 0.1 | −6.3 ± 0.1 | −49.6 ± 0.2 | −9.6 |
| 4c | −47.8 ± 0.1 | 98.8 ± 0.5 | −74.9 ± 0.3 | −6.2 ± 0.1 | −30.1 ± 0.2 | −5.0 |
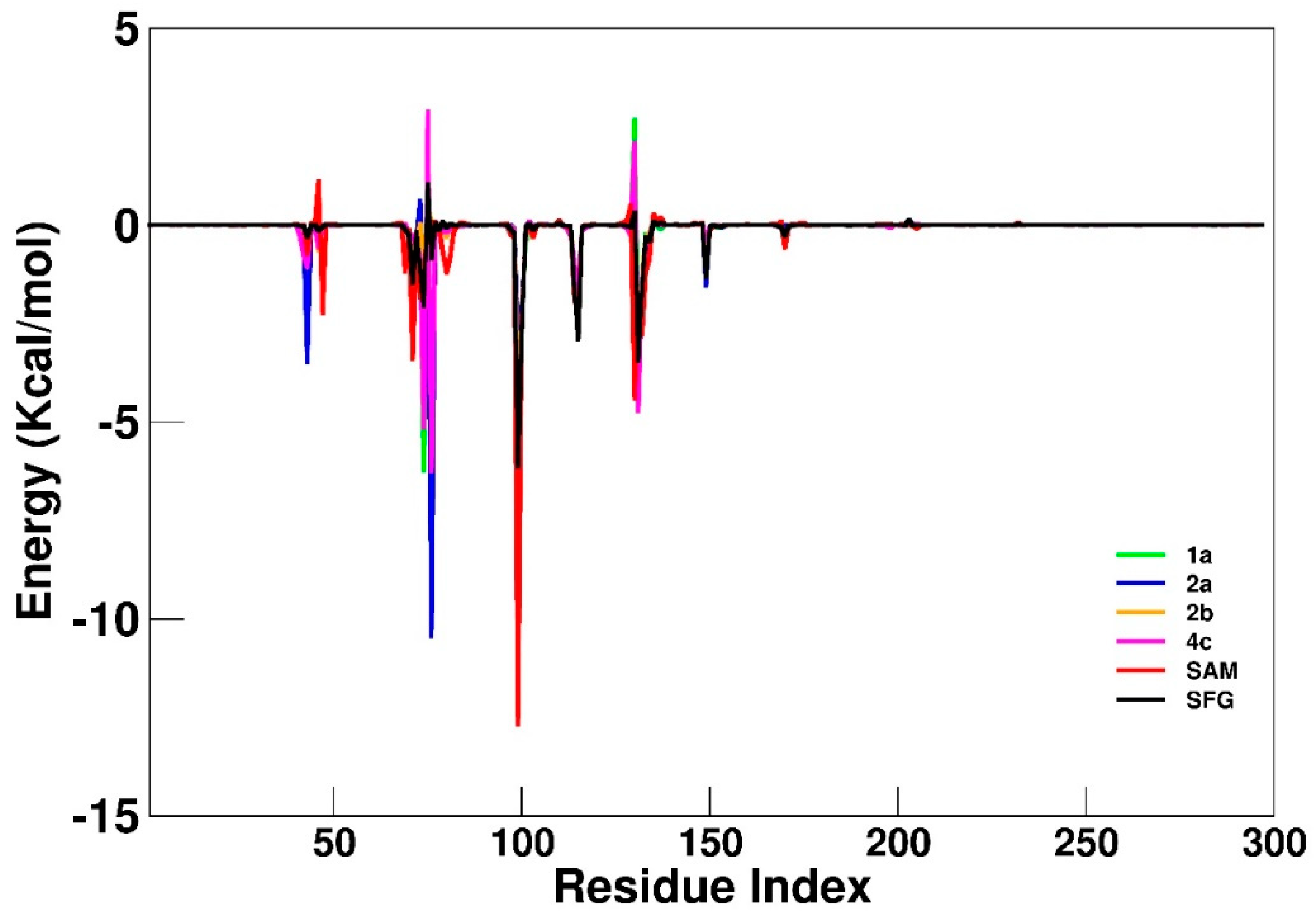
| AA Residue | SAM | SFG | 1a | 2a | 2b | 4c |
|---|---|---|---|---|---|---|
| Asn43 | −0.7 | −0.3 | −3.4 | −3.5 | −0.9 | −1.1 |
| Tyr47 | −2.2 | 0.0 | 0.0 | 0.0 | −0.2 | −0.1 |
| Gly73 | −1.8 | −1.2 | 0.0 | 0.6 | 0.0 | −0.8 |
| Ser74 | −1.0 | −2.0 | −6.2 | −4.8 | −1.0 | −5.1 |
| Lys76 | 0.1 | −0.9 | −4.3 | −10.4 | 0.1 | −6.2 |
| Asp99 | −12.7 | −6.1 | −5.2 | −2.6 | −4.0 | −8.2 |
| Leu100 | −2.6 | −2.9 | −2.3 | −2.3 | −2.6 | −2.6 |
| Asp114 | −1.8 | −1.6 | −1.3 | −1.2 | −1.0 | −1.0 |
| Cys115 | −2.5 | −2.9 | −2.7 | −2.5 | −2.4 | −1.6 |
| Asp130 | −4.4 | 0.3 | 2.7 | 0.7 | 1.2 | 2.1 |
| Met131 | −2.4 | −3.5 | −3.0 | −2.9 | −4.1 | −4.7 |
| Tyr132 | −2.8 | −1.6 | −1.3 | −2.2 | −1.2 | −1.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balieiro, A.M.; Anunciação, E.L.S.; Costa, C.H.S.; Qayed, W.S.; Silva, J.R.A. Computational Analysis of SAM Analogs as Methyltransferase Inhibitors of nsp16/nsp10 Complex from SARS-CoV-2. Int. J. Mol. Sci. 2022, 23, 13972. https://doi.org/10.3390/ijms232213972
Balieiro AM, Anunciação ELS, Costa CHS, Qayed WS, Silva JRA. Computational Analysis of SAM Analogs as Methyltransferase Inhibitors of nsp16/nsp10 Complex from SARS-CoV-2. International Journal of Molecular Sciences. 2022; 23(22):13972. https://doi.org/10.3390/ijms232213972
Chicago/Turabian StyleBalieiro, Alessandra M., Eduarda L. S. Anunciação, Clauber H. S. Costa, Wesam S. Qayed, and José Rogério A. Silva. 2022. "Computational Analysis of SAM Analogs as Methyltransferase Inhibitors of nsp16/nsp10 Complex from SARS-CoV-2" International Journal of Molecular Sciences 23, no. 22: 13972. https://doi.org/10.3390/ijms232213972