1. 引言
2020年12月中旬,英国首次报告了传染性极强的新冠(SARS-CoV-2)病毒突变株:B.1.1.7毒株,这一毒株很快成为伦敦地区主要毒株。
S蛋白(spike protein,刺突蛋白)在冠状病毒进入宿主细胞时,主要由跨膜刺突(S)糖蛋白进行介导并参与入胞过程,在新型冠状病毒的传播过程中发挥着重要的作用 [1]。在研究新冠病毒基因序列时,S蛋白表位也一直是抗体研发的研究对象,它具有一定的代表性和特征性 [2],所以主要以分析S蛋白为出发点来进行进一步的研究 [3]。而B.1.1.7毒株在S蛋白受体结合结构域(RBD) 501位置出现突变,这让我们将目光放到了B.1.1.7毒株的S蛋白上。
目前,基因研究的常规方式是通过基因特征提取、基因序列定位等方式来找到关键位点进行研究 [4]。
信息熵1的概念自1948年被香农提出后,随着科技的发展,已突破香农信息论的范围,在生物医学领域被研究应用并取得成果,成为现代生物医学领域中的一种新思路、新方法。因此本文将信息熵引入生物信息学领域,根据信息熵的概念和香农公式 [2],结合生物学领域的计算和分析要求,对需要的生物信息进行熵值计算和分析,之后将计算出的熵值在二维空间进行投影,以达到可视化分析的目的 [3] [5] [6] [7]。
本文将在第二章中介绍系统架构,使用S蛋白熵值可视化将S蛋白的基因序列进行处理、计算、投影成二维可视化图示,在第三章中以可视化的结果来对复杂的S蛋白进行展示。
2. 系统架构
2.1. 参数解释
m:DNA序列各分组长度(这里我们选取了m = 80进行可视化处理分析);
M:每条序列组数,
(ALL为每条序列总碱基数);
:每组分组对应排列N (
;N = A、C、G、T、AC、AG、AT、CG、CT、GT、ACG、
ACT、AGT、CGT、ACGT,顺序无关)的出现概率,
;
:每条序列各排列分组的信息熵(N = A、C、G、T、AC、AG、AT、CG、CT、GT、ACG、ACT、AGT、CGT、ACGT),计算公式为
:每条序列信息熵映射生成图像上的点,如
即表示该条序列碱基A与C的信息熵在图像上的映射。
2.2. 架构
系统整体架构如图1所示,分为输入、计数、处理、投影、输出五个模块,其中核心功能模块有三个,分别是处理、测量、投影模块。
计数模块的功能是将新冠病毒基因序列S蛋白以m个碱基为一段,进行四种碱基(A,T,C,G)的数量统计;在处理模块中需要对各段信息熵进行计算,再累加各段信息熵得到S蛋白的熵值;在投影模块中,可以选择查看单张信息熵分布图或225张信息熵分布图的全排列。
2.3. 计数模块
计数模块是针对输入的编码S蛋白的基因序列进行处理,下载的编码S蛋白的基因序列自动以80个碱基为一段分好,不需要手动分段,因此直接导入计数程序即可计算出每一段中四种碱基(A,T,C,G)的数量,架构图如图2所示。
2.4. 处理模块
处理模块主要是根据香农公式对计数模块输出的结果进行处理,分别计算出整条编码S蛋白基因的15种碱基组合(N = A、C、G、T、AC、AG、AT、CG、CT、GT、ACG、ACT、AGT、CGT、ACGT)的熵值 [5] [6],架构如图3所示。
2.5. 投影模块
投影模块可以将除含空集外的15个碱基组合中的任意两个的信息熵值作为X、Y轴,生成相关的信息熵分布图像,并使用不同颜色加以区分,也可以直接生成225张散点图,以N中排列顺序输出到同一张大图中,架构如图4所示。
3. 过程与结果分析
3.1. 序列选择
本文意图通过研究新冠病毒变异毒株S蛋白与初期毒株的S蛋白信息熵的差异,找出毒株突变的生物学原理与生物学表达,为生物信息、生命科学等方面提供一定的研究基础,因此我们选择了B.1.1.7毒株中的四条序列和原始序列,提取S蛋白段进行尝试。
3.2. 序列比对
选取初期序列(NC_045512.2)作为基准序列和B.1.1.7毒株棘突蛋白的四条序列(三条来自苏格兰的序列CVR5974、CVR6031、CVR6032,一条来自英格兰的序列204590575),使用对齐工具MEGA6对齐五条序列,根据S蛋白的前后碱基排列特征截取五条序列的S蛋白段。
对病毒基因序列的S蛋白段进行碱基对比分析,如图5所示。
从图中信息可以看出自英格兰的病毒序列包含较多N碱基,取自苏格兰的三条病毒序列相似度较高,变异毒株的S蛋白与初期序列均有差异。
3.3. 可视化分析
如图6为各碱基组合(N = A、C、G、T、AC、AG、AT、CG、CT、GT、ACG、ACT、AGT、CGT、ACGT)对应的信息熵全映射。从图中我们清晰直观地可以看出,五条序列的碱基信息熵区间均在3到4之间,成聚集的情况,而且均存在差别,这表明了突变的发生。
如图7所示,可以选取任意目标碱基组合放大,对图像映射进行更有效的处理分析。
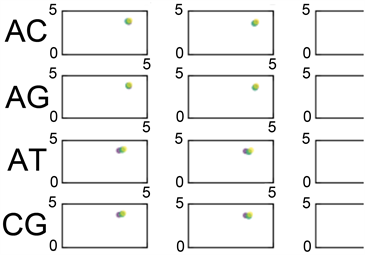
Figure 7. Zooming in on target pairs mapping
图7. 放大目标组合映射
此处我们选取了A、C、AT、CG组合进行细致的规律可视化分析,从图8~11中看出,来自苏格兰的三条序列CVR5974、CVR6031、CVR6032的信息熵映射呈集聚之势,可以推测三者之间的亲属关系,且相对于另外的两条序列的映射,在表现上具有不稳定性。而参考序列NC_045512.2与英格兰的序列204590575在趋势上呈现不变性,但明显可以区别参考序列、苏格兰三条序列与英格兰一条序列之间的关系。突变的一部分特性在图像上得以体现。
S蛋白个别位点的变化就有可能会造成整个病毒的变异,在病毒功能性中起到关键作用。信息熵分布图的差异性就取决于样本的差异性,相同的S蛋白样本只会产生一个特征点。因此通过信息熵分布图像,可以批量导入S蛋白段后放大观察,选取有差异的基因序列继续细化观察,通过全排列的图像可以基本发现出现差异的碱基变化,从而获取到一些特征信息,再配合其他工具细化分析。
同时,信息熵可视化作为一种分析方法,可以扩展应用到一切病毒的研究分析中,包括整段的基因序列分析,有着广泛的应用前景。
4. 总结
本文通过对新型冠状病毒变异毒株和原始病毒进行信息熵计算及投影,在实现过程中可以根据需求调整每组碱基数量,将数据转换为更加直观的彩色散点图,根据需要放大局部并进行分析。相比于传统的生物研究方法,信息熵投影可以提高数据分析效率,为生物信息、计算生物等方面研究提供了新思路和研究基础。
致谢
感谢郑智捷教授的悉心指导,感谢云南大学软件学院对本项目的支持。
基金项目
国家自然科学基金项目62041213。
NOTES
1信息熵:香农从热力学中借用过来,以描述信源的不确定度。