
Abstract
The prevalence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) among polycystic ovary syndrome (PCOS) is significantly higher than in the general population. However, the mechanisms underlying this remain obscure. This study aimed to explore the mechanisms by identifying the genetic signature of SARS-CoV-2 infection in PCOS. In the present study, a total of 27 common differentially expressed genes (DEGs) were selected for subsequent analyses. Functional analyses showed that immunity and hormone-related pathways collectively participated in the development and progression of PCOS and SARS-CoV-2 infection. Under these, 7 significant hub genes were identified, including S100A9, MMP9, TLR2, THBD, ITGB2, ICAM1, and CD86 by using the algorithm in Cytoscape. Furthermore, hub gene expression was confirmed in the validation set, PCOS clinical samples, and mouse model. Immune microenvironment analysis with the CIBERSORTx database demonstrated that the hub genes were significantly correlated with T cells, dendritic cells, mast cells, B cells, NK cells, and eosinophils and positively correlated with immune scores. Among the hub genes, S100A9, MMP9, THBD, ITGB2, CD86, and ICAM1 demonstrated potential as possible diagnostic markers for COVID-19 and PCOS. In addition, we established the interaction networks of ovary-specific genes, transcription factors, miRNAs, drugs, and chemical compounds with hub genes with NetworkAnalyst. This work uncovered the common pathogenesis and genetic signature of PCOS and SARS-CoV-2 infection, which might provide a theoretical basis and innovative ideas for further mechanistic research and drug discovery of the comorbidity of the two diseases.
Similar content being viewed by others

Introduction
Recently, several countries and regions have been experiencing outbreaks of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) since late 20191. Infection with SARS-CoV-2 leads to Coronavirus disease 2019 (COVID-19), which can be asymptomatic or presents as acute respiratory distress syndrome2. However, in multiple large observations, the virus also readily infected extrapulmonary organs, including the heart, eyes, and kidneys. People with diabetes, obesity, and cardiovascular disease are more likely to experience severe symptoms and die from COVID-193. Therefore, preventive strategies are warranted to protect persons with comorbidities from developing severe COVID-194.
Men have been reported to be more prone to contracting COVID-19 than women possibly due to a complex interplay of biological, hormonal, and immunological factors5,6,7,8. For instance, estrogen is known for its immunomodulatory effects, potentially offering protective benefits against viral infections in women9,10. Recent evidence suggests that hormonal regulation of TMPRSS2 expression, another critical protein involved in viral cell entry, further underscores the differential susceptibility between males and females11. These insights highlight the importance of considering hormonal profiles in assessing COVID-19 outcomes, particularly in the context of sex-specific vulnerabilities.
Polycystic ovary syndrome (PCOS), the most common endocrine disorder, affects about 5–15% of women of reproductive age and is characterized by hyperandrogenism, oligoovulation, and polycystic ovaries12. It was reported that more than 80% of PCOS patients suffer from hyperandrogenemia13. Androgens, including testosterone, have been implicated in exacerbating COVID-19 severity by upregulating the ACE2 receptor, which facilitates viral entry into host cells14. In addition, women with PCOS were at risk of long-term complications, including obesity, type 2 diabetes mellitus, nonalcoholic fatty liver disease, and cardiovascular disorders15. Additionally, a recent epidemiological study revealed that women with PCOS were remarkably more likely than controls to contract COVID-1916. These studies suggest that PCOS patients are at higher risk of COVID-19. Considering PCOS patients with COVID-19 are at an elevated risk for complications, discerning synergistic biomolecular pathways is crucial for identifying therapeutic targets and repurposing medicines for PCOS patients with COVID-19. With the rapid development of sequencing technology, transcriptome analysis has been widely applied to study the pathogenesis of PCOS and COVID-1917,18. However, the link between PCOS and vulnerability to COVID-19 remains uncertain.
In this study, we used bioinformatics methods to screen common differentially expressed genes (DEGs) of COVID-19 and PCOS, determine their signaling pathways and immune microenvironment, and build the interaction networks with miRNAs, transcript factors, drugs, and chemical compounds. The research may provide new perspectives and strategies for treating PCOS patients infected with SARS-CoV-2.
Materials and methods
Acquisition of datasets
Gene Expression Omnibus (GEO) was used to obtain all eligible datasets. We used four datasets for this study, including three PCOS (GSE34526, GSE10946, and GSE54248) and three SARS-CoV-2 datasets (GSE147507, GSE157103 and GSE164805). GSE34526 and GSE147507 were derived from expression profiles using array, while GSE157103 and GSE10946 were derived from expression profiles using high-throughput sequencing. We selected normal and SARS-CoV-2 infected lung epithelial cell NHBE as part of the analysis of the GSE147507 dataset. In addition, 40 female COVID-19 and 12 female non-COVID-19 samples from the GSE157103 dataset were selected for this study. PCOS clinical samples and mouse models were used to analyze the identified hub genes. Details of the dataset are shown in Table 1.
Ethics approval
The clinical samples were from the previous collection and described in an earlier publication19. The research followed the principles of the Declaration of Helsinki and was approved by the local Human Investigation Ethics Committee (license numbers APK.002.9.2020 and APK.002.38.2021). Written informed consent was obtained from all patients before inclusion. All animal experiments were approved by the Ethics Committee for Animal Experimentation of Xuzhou Medical University (license number 202209S077). C57BL/6J mice were obtained from Beijing Vital River Laboratory Animal Technology. We confirm that all methods were carried out in accordance with relevant guidelines and regulations and that all methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Identification of DEGs and Enrichment terms
The datasets were downloaded from GEO, normalized, and DEGs were extracted with the R package “edgeR”20. Fold changes (FCs) were calculated for individual genes. DEGs were defined using a p < 0.05 and |logFC|>1.0. The R language package Venn Diagram was used to obtain shared DEGs21. Heatmap and volcano plots were created using an online platform (https://www.bioinformatics.com.cn). GO and KEGG enrichment analysis was performed using R, and adjusted p < 0.05 was used to select enriched terms22,23.
Protein-protein interactions (PPI) network construction
The PPI interaction network was constructed by STRING ( http://string-db.org)24 and visualized by Cytoscape (version 3.9.1)25. Molecular Complex Detection (MCODE) in Cytoscape was used to analyze core functional modules26. CytoHubba, a plugin for Cytoscape, was used to select hub genes. Seven algorithms were applied to confirm the final hub genes, which were then visualized with Venn diagrams27. Finally, we used Gene MANIA (www.genemania.org) to create a co-expression network of hub genes28.
Gene set enrichment analysis (GSEA) analysis
We obtained the GSEA software (version 3.0) from the GSEA website and divided the samples into high ( ≥ 0%) and low (< 50%) expression groups based on gene expression levels29. The “cluster Profiler” package was used to perform GSEA on potential mechanisms of c2 (c2. cp.v7.5.1. symbols.gmt) in the Molecular Signature Database (MSigDB)30. Pathways with false discovery rates (FDR) less than 0.05 were considered significant.
Immune microenvironment analysis
Datasets GSE34526 and GSE157103 were downloaded from the GEO database for data pooling. In order to obtain individual gene immune scores and expression matrices for 22 immune cells, CIBERSORTx was used to analyze the immune microenvironment of human immune cells with the corresponding genes S100A9, MMP9, TLR2, THBD, ITGB2, ICAM1, CD8631. The scatter density of each gene was plotted using the “ggplot2” package in R.
Genes, drugs, and chemical compounds-hub genes regulatory networks establishment
We established hub gene-miRNA, hub gene-transcription factor (TF), hub gene-drug, and hub gene-chemical compound interaction network using NetworkAnalyst32. A hub gene-miRNA interaction network was discovered using TarBase and miRTarBase databases. In addition, a TF-DEG interaction network was developed using the JASPAR database33.
PCOS patient sample collection and mouse model built
The details of the granulosa cell collection and PCOS mouse model construction were performed as previously described publication19. For the collection of granulosa cells, in brief, the follicular fluid was centrifuged at 250×g for 10 min, and the granulosa cells were aspirated and placed in a new centrifuge tube. The cells were washed with PBS, resuspended, and collected by centrifugation again.
For the PCOS mouse model, in brief, 3-week-old female C57BL/6J mice were injected subcutaneously with DHEA (6 mg/100 g body weight) or an equivalent dose of sesame oil for 28 consecutive days, and their ovarian tissue was collected for assay analysis.
All animal procedures were conducted in accordance with the a method approved by our institution’s Animal Care and Use Committee (ACUC). We used a humane and scientifically validated method for euthanasia, which involved administering an overdose of isoflurane. This approach ensured that the procedure was carried out with minimal distress to the animals. Following the induction of deep anesthesia, the mice were euthanized through cervical dislocation, a method recommended for its efficiency and humane qualities.
Hub gene testing and receiver operating characteristic curve (ROC) curve plotting
Hub genes testing was performed as previously described34. Briefly, RNA was extracted from mouse ovary tissues or granulosa cells from Control, and PCOS patients using the TRIzol method and reverse transcribed using a cDNA kit. Hub gene expression was detected by qPCR, and glyceraldehyde phosphate dehydrogenase (GAPDH) was used as an internal reference. Primer sequences are listed in Table S1.
Using the R package “pROC”, we constructed ROC curves and calculated the area under the ROC curves (AUC) for each hub gene to assess its diagnostic performance35.
Statistical analysis
Values were analyzed by the two-tailed Student’s t-test with the GraphPad Prism 8.0.1 software and presented as mean ± standard error of the mean (SEM). The significance of the statistics is expressed by the value of p. *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Differentially expressed genes (DEGs) identification
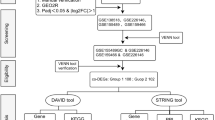
The research content and ideas of this study are shown in Fig. 1. We used the SARS-CoV-2 dataset GSE147507 and PCOS dataset GSE34526 from the Gene Expression Omnibus (GEO) database to analyze DEGs of COVID-19 and PCOS. Differential expression genes were collected from primary human lung epithelium (NHBE) cells infected with SARS-CoV-2 and granulosa cells of PCOS patients. After screening, DEGs were identified with a p < 0.05 and |log2 FC|>1.0 and visualized using a volcano plot and heatmap (Fig. 2A and B).

The research content and ideas of this study are shown in Fig. 1.

Differential expression genes (DEGs) and enrichment pathway identification. (A) Volcano plot showed the DEGs in GSE147507 (SARS-Cov-2 dataset). (B) Volcano plot showed the DEGs in GSE34526 (PCOS dataset). (C) Venn diagram displayed 27 overlapping DECs in the two GSE sets. (D and E) GO and KEGG enrichment analysis of overlapping DEGs. GO analysis showing the biological, cell component and molecular function enrichment results (D). KEGG analysis showing the signaling pathway enrichment results (E). An adjusted p < 0.05 was identified as significantly changed. GO gene ontology, BP biological process, CC cellular component, MF molecular function, KEGG Kyoto encyclopedia of genes and genomes.
Overlapping gene and enrichment pathway screening
Based on a Venn diagram analysis, 27 common DEGs overlapped between GSE34526 and G147507 (Fig. 2C). The expression levels of the 27 genes are shown in Supplementary Fig. (Figure) 1 and Fig. S2. We used the GO and KEGG databases to conduct pathway enrichment analyses to understand better the biological functions of the DEGs identified. We found that DEGs contributed to biological processes, including inflammatory response, reaction oxygen species metabolic process, and NF-κB transcription factor activity (Fig. 2D). In addition, DEGs were associated with molecular functions such as adenylyltransferase activity, toll-like receptor binding, and RAGE receptor binding (Fig. 2D). Similarly, KEGG was enriched in the IL17 signaling pathway, virus infection, cell adhesion molecules, phagosome, TNF signaling pathway, and NOD-like receptor signaling pathway (Fig. 2E).
Differential expression genes and hub gene interaction network construction
The interaction between DEGs was analyzed using the PPI network based on the STRING database and visualized using Cytoscape (Fig. 3A). The MCODE plugin of Cytoscape was used to obtain two gene modules, including 12 shared DEGs (Fig. 3B and C). The top seven hub genes, including TLR2, MMP9, S100A9, ICAM1, THBD, ITGB2, and CD86, were acquired with Venn diagrams via the seven algorithms in CytoHubba (Fig. 3D). We built a complex gene interaction network per the GeneMANIA database (Fig. 3E). Based on the GO analysis of the hub gene, we found that the hub genes were primarily responsible for biological functions such as response to hormones, phagocytosis, viral life cycle, response to insulin, interaction with the host, and response to interferon-gamma (Fig. 3F). Moreover, the major enrichment pathways in the KEGG analysis were related to virus infection, toll-like receptor signaling pathway, phagosome, and TNF signaling pathway (Fig. 3G).

Figure 3 DEGs and hub gene interaction network construction. (A) Analysis results of PPI network in STRING online tool and visualization with Cytoscape. (B and C) Two significant gene clustering modules were analyzed with Cytoscape. (D) Venn diagram displayed that 7 overlapping hub genes were screened out with 7 algorithms. (E) Hub genes and their co-expression genes were analyzed via GeneMANIA. (F) and (G) GO and KEGG enrichment analysis of the hub genes. The left side of the figure is a sankey diagram, which represents the genes contained in each pathway, and the right side is a conventional bubble diagram. The size of the bubble indicates the number of genes to which the pathway belongs, and the color of the bubble indicates the p value.
GSEA analysis of hub genes
We used GSEA enrichment to perform systematic KEGG pathway mining for each of the seven hub genes. The TOP 6 signaling pathways for each gene are illustrated in Fig. 4. We discovered that the hub genes were significantly enriched to amino acids, glycans metabolism, and immune-related pathways, suggesting that immunity and metabolism may play a key role in PCOS and SARS-Cov-2 infection.

GSEA analysis of hub genes. GSEA gene set enrichment analysis.
Immune microenvironment analysis of hub genes
To explore the relationship between hub genes and the immune microenvironment, we analyzed the level of the immune microenvironment of hub genes S100A9, MMP9, TLR2, THBD, ITGB2, ICAM1, CD86 using the tool CIBERSORTx. The results revealed that 7 hub genes were significantly correlated with T cells, dendritic cells, mast cells, B cells, NK cells, and eosinophils among the 22 types of immune cells (Fig. 5A) and positively correlated with immune scores (Fig. 5B and H). These results suggest that the hub genes were significantly correlated with the infiltration of immune cells.

Immune microenvironment analysis. (A) Correlation between hub gene expression levels and 22 immune cells. (B–H) Correlation analysis of hub gene expression levels with immune scores.
Interaction analysis of hub genes with ovary-specific genes, miRNAs, transcription factor (TF), and drugs
We investigated the association of ovary-specific genes and pathways with hub genes. As shown in Fig. 6A, the yellow and blue circles represent hub genes and ovary-specific genes, respectively. These genes had biological functions related to immunity, ovarian follicle formation, and the ovulation cycle (Fig. 6B). Furthermore, KEGG pathways were enriched in leukocyte transendothelial migration, toll-like receptor signaling, and GnRH signaling pathways (Fig. 6C). We used the TarBase and miRTarBase databases to analyze hub gene-miRNA interactions. A network of interactions is shown in Fig. 7A. Among them, the expression of three node miRNAs, miR-146a-5p, miR-335-5p, and miR-21-5p, was significantly downregulated in clinical samples and mouse models of PCOS (Fig. 7B and C). In addition, we identified the interaction network between the TF and hub genes with the JASPER database (Fig. 8A) and drugs on hub genes (Fig. 8B). The information on hub gene and drug relationship was obtained from the Drug Bank database. The drugs found in our analysis include resveratrol, methotrexate, nickel, tretinoin, progesterone, and estradiol.

Hub genes and ovary specific genes interaction network analysis. (A) Hub genes-ovary specific genes interaction network. Yellow dots represent hub genes and blue dots represent transcription factors. (B,C) GO (B) and KEGG (C) enrichment analysis of the interacting genes.

Hub gene-microRNA interaction network analysis. (A) Hub gene-microRNA interaction network. Hub genes were depicted as red circles, while miRNAs were represented as blue squares. (B) Expression of node miRNAs in PCOS clinical samples. (C) Expression of node miRNAs in the ovaries of PCOS mice. *P<0.05, **P<0.01.

Hub gene-transcription factors (TFs), hub genes-drugs, hub genes–chemical compounds interaction network analysis. (A) Hub gene-TFs interaction network analysis. The red circle and green square represented the hub genes and the TFs, respectively. A node’s size was determined by its degree. (B) Hub genes-drugs interaction network analysis. Drugs and hub genes represented by blue squares and green circles.
Hub gene expression and ROC curve plotting
To clarify the expression of hub genes in SARS-Cov-2 and PCOS samples, we assessed their expression in training sets GSE147507 for SARS-CoV-2 infection and GSE34526 for PCOS (Fig. 9). All hub genes were upregulated in SARS-CoV-2 and PCOS compared to the control group. Moreover, we confirm their expression in validation sets GSE15703 and GSE10946 (Fig. 10A and N). Consistent with the training set results, our findings demonstrated elevated expression of hub genes in both COVID-19 and PCOS samples relative to controls. Blood transcriptome analysis revealed predominant upregulation of these hub genes except ITGB2 in peripheral blood from COVID-19 and PCOS patients, aligning generally with the observations above (Fig. 10O and P). More importantly, we collected clinical samples and constructed a PCOS mouse model for further validation. The results exhibited that the hub genes were significantly higher in the granulosa cells from PCOS patients and ovarian tissues of PCOS mice compared with controls (Fig. 11). In general, these results were in line with the above analysis.

Expression levels of hub genes in training sets. (A–G) Expression levels of hub genes in training set GSE147507. (H–N) N Expression levels of hub genes in training set GSE34526. *P<0.05, **P<0.01, ***P<0.001.

Expression levels of hub genes in validation sets. (A–G) Expression levels of hub genes in validation set GSE157103. (H–N) Expression levels of hub genes in validation set GSE10946. (O) and (P) Expression levels of hub genes in the peripheral blood datasets GSE 164,805 (COVID-19) and GSE54248 (PCOS). *P<0.05, **P<0.01, ***P<0.001.

Expression levels of hub genes in PCOS clinical samples and mouse model. (A–G) Expression levels of hub genes in PCOS clinical samples, n = 20. (H–N) Expression levels of hub genes in PCOS mouse model, n = 6. *P<0.05; **P<0.01; ***P<0.001.
Based on the validation sets, we evaluated the diagnostic performance of 7 hub genes in COVID-19 and PCOS utilizing ROC curves. The results showed that ITGB2, CD86, S100A9, THBP, and MMP9 exhibited potential diagnostic performance, while TLR2 had poor diagnostic performance (Fig. 12).

Receiver operator characteristic curve (ROC) diagnostic curve of hub genes in validation set GSE157103 and GSE10946.
Discussion
Several retrospective studies reported PCOS as one of the most common comorbidities of COVID-1936. However, why PCOS patients are susceptible to COVID-19 remains uncertain. Therefore, understanding the molecular mechanisms, early diagnosis, and intervention is of great clinical importance. In this work, we performed bioinformatics analyses on two independent gene chip databases containing SARS-CoV-2 infected NHBE and PCOS granulosa cells and identified 27 common DEGs.
According to GO, KEGG, and GSEA enrichment analysis, the DEGs were enriched primarily in immune response, NF-kappaB transcription factor activity regulation, Toll-like receptor (TLR) binding, virus infection, TNF signaling pathway, IL-17 signaling pathway, and NOD-like receptor signaling pathway. These findings suggest that COVID-19 and PCOS have DEGs associated with inflammation and immune response. It has also been reported that severe COVID-19 triggers an exaggerated inflammatory response that can lead to acute respiratory distress syndrome, a life-threatening condition associated with multi-organ failure and high mortality37. By Pearson correlation analysis, we found that 7 hub genes were significantly positively correlated with immune scores, implying that they are associated with immune infiltration. Hub gene expression heatmaps showed that hub gene expression was low in innate immune cells such as monocytes, activated NK cells, and neutrophils. Most of the hub genes had lower expression in M2 macrophages, whereas they were highly expressed in M1 macrophages. As is well known, M2 macrophages suppress inflammation while M1 macrophages promote it38. The differential expression of hub genes in macrophages may be a common pathogenic cause of PCOS and SRAS-Cov-2. Furthermore, in the training set, validation set, animal models, and clinical samples, the expression of hub genes was significantly higher in the PCOS and SARS-Cov-2 groups than in the control group. This suggests that high expression of these genes is associated with the development of inflammation. Recent findings have indicated that the periovulatory follicles of women with PCOS contain elevated inflammatory mediators and that this low-grade inflammation might serve as a precursor to ovarian dysfunction in PCOS39. This underlying proinflammatory predisposition may put women with PCOS at increased risk of severe COVID-1914.
Based on the MCODE and seven algorithms of CytoHubba, we screened seven hub genes, including S100A9, TLR2, CD86, ICAM, THBD, MMP9, and ITGB2. This study will focus on the effects of these hub genes on PCOS and COVID-19 from an inflammatory perspective. TLR2 was a key hub gene among the hub genes. During a viral infection, the host uses pattern recognition receptors, especially TLRs, to sense the virus and activate the innate immune system40. TLR2 was thought to be one of the most significant members of the TLR family and is responsible for sustaining airway inflammation41. In line with this, GO, and KEGG Pathway identified significant enrichment in signaling pathways involving toll-like receptors. In addition, COVID-19 severity was associated with TLR2 expression. Coronavirus infection caused proinflammatory cytokines to be produced independently of viral entry via TLR2-dependent signaling. TLR2 sensed the SARS-CoV-2 envelope protein as its ligand42,43. Besides, there was evidence that TLRs are located in ovary granulosa cells, cumulus cells, and theca cells44. The abnormal expression of TLRs did not result in good oocyte quality and insufficient fertility45. In another study, TLR2 was found to be highly expressed in granulosa cells from PCOS patients and could mediate inflammation and oxidative stress caused by LPS46. These results showed that TLR2 was a key molecule mediating inflammation in COVID-19 and PCOS. We speculate that the state of chronic inflammation in PCOS primarily mediates the susceptibility of affected patients to COVID-19. Other hub genes were also associated with inflammatory pathways or metabolic pathways, which were involved in PCOS and became potential risk factors for susceptibility to COVID-1947,48,49.
S100A9, also known as calgranulin B, is a proinflammatory protein implicated in various inflammatory disorders50. S100A9 is posited to exacerbate the inflammatory state within this disorder, intensifying metabolic dysfunction and reproductive anomalies51,52. Elevated S100A9 levels have been associated with increased adipose tissue inflammation and insulin resistance—hallmarks of PCOS53. Meanwhile, S100A9 was found to be upregulated in PCOS samples by dataset analysis, clinical samples, and animal model assays in this study. These findings suggest that S100A9 not only contributes to the inflammatory environment but may also serve as a therapeutic target to alleviate the metabolic and reproductive complications of PCOS. In the context of COVID-19, S100A9 appears to play a detrimental role by intensifying inflammatory responses, potentially leading to severe lung injury and systemic complications. The protein’s overexpression in severe COVID-19 cases correlates with elevated cytokine levels, enhanced neutrophil infiltration, and worse clinical outcomes50,54. These observations indicate that S100A9 could act both as a biomarker of disease severity and as a target for therapeutic intervention to modulate the inflammatory response in COVID-19 patients. Specifically, women with PCOS who contract COVID-19 could face more severe manifestations due to pre-existing chronic inflammation and metabolic dysregulation, potentially mediated by elevated S100A9 levels. Trials exploring inhibitors of S100A9 or its signaling pathways could elucidate strategies to reduce the inflammatory burden in these patients, potentially improving symptoms and outcomes for both PCOS and COVID-19.
CD86, also known as B7-2 or B70, plays a pivotal role as a co-stimulatory molecule involved in immune regulation, particularly in antigen presentation and T-cell activation55. CD86 is prominently expressed on antigen-presenting cells such as dendritic cells and macrophages, which are crucial for activating T cells via interactions with CD2856. This interaction perpetuates inflammatory responses observed in PCOS, potentially exacerbating insulin resistance and ovarian dysfunction. Elevated CD86 expression correlates with conditions marked by chronic inflammation, underscoring its likely involvement in the pathogenesis of PCOS57. Consistent with this, this study also found elevated expression of CD86 in PCOS samples. In the context of COVID-19, CD86 is implicated in the dysregulated immune responses observed in severe cases of the disease. Dysregulated immune activation, including excessive co-stimulatory signaling pathways like CD86/CD28 interactions, contributes to the cytokine storm syndrome and subsequent tissue damage characteristic of severe COVID-19, particularly affecting pulmonary and vascular systems58. The convergence of CD86-mediated inflammatory pathways in both PCOS and COVID-19 suggests synergistic effects in individuals affected by both conditions. Exploration of CD86-targeted therapeutic strategies offers novel avenues to mitigate inflammation-driven complications in these conditions.
ICAM-1, a pivotal cell surface glycoprotein central to immune responses and leukocyte adhesion59.ICAM-1, expressed on endothelial cells and leukocytes, facilitates immune cell adhesion and transmigration across endothelial barriers, thereby exacerbating localized inflammation and insulin resistance. Elevated ICAM-1 levels have been observed in PCOS patients, correlating with markers of inflammation and insulin resistance, suggesting a contributory role in the pathophysiology of metabolic and reproductive dysfunctions associated with PCOS48. Severe COVID-19 cases frequently exhibit endothelial activation and dysfunction, leading to enhanced vascular permeability and immune cell infiltration in vital organs. Upregulated ICAM-1 expression promotes leukocyte recruitment and adherence to endothelial cells, contributing to the cytokine storm syndrome and tissue damage characteristic of severe disease60. Higher ICAM-1 levels correlate with disease severity and adverse clinical outcomes in COVID-19 patients, highlighting its significance as a biomarker and potential therapeutic target61. The convergence of inflammatory pathways involving ICAM-1 in both PCOS and COVID-19 suggests a synergistic impact in individuals affected by both conditions. Women with PCOS contracting COVID-19 may experience heightened inflammatory responses and endothelial dysfunction, potentially exacerbating metabolic and cardiovascular complications linked to PCOS; nonetheless, how ICAM1 affects PCOS and SARS-Cov-2 needs further exploration.
Thrombomodulin (THBD) is a pivotal glycoprotein renowned for its multifaceted roles in coagulation regulation and endothelial function62. THBD, primarily expressed on endothelial cells, plays a crucial role in modulating coagulation dynamics by converting thrombin to its anticoagulant form. It possesses anti-inflammatory properties through interactions with protein C63. SARS-CoV-2 infection triggers systemic inflammation, endothelial cell activation, and microvascular thrombosis, contributing significantly to disease severity64. THBD’s anticoagulant properties are pivotal in maintaining vascular integrity by inhibiting thrombin-mediated coagulation and attenuating endothelial activation65. However, in severe COVID-19 cases, endothelial cell shedding of THBD compromises its protective functions, exacerbating coagulopathy and inflammatory responses66. The dual functionality of THBD in regulating coagulation and inflammation underscores its potential impact on disease outcomes in both PCOS and COVID-19. Thus, comprehending the intricate involvement of THBD in these conditions is crucial for developing targeted therapeutic strategies aimed at mitigating vascular and inflammatory complications.
MMP9 plays a pivotal role in extracellular matrix remodeling and inflammation, implicating its involvement in PCOS and COVID-19 pathophysiology67. Elevated MMP9 levels correlate with disrupted ovarian follicular development, impaired ovulation, and insulin signaling abnormalities observed in PCOS patients68. This is consistent with the results obtained in this study. These mechanisms underscore MMP9’s integral role in metabolic dysfunction and reproductive disturbances associated with the syndrome. Conversely, in COVID-19, MMP9’s activity exacerbates inflammatory responses critical to disease severity and outcomes69. Particularly in severe cases marked by cytokine storms and acute respiratory distress syndrome, MMP9 promotes immune cell infiltration and endothelial barrier disruption, contributing to lung injury and systemic inflammation70. Elevated MMP9 expression correlates with increased pulmonary fibrosis and adverse clinical outcomes in COVID-19 patients, highlighting its potential as both a prognostic biomarker and therapeutic target71. Understanding MMP9’s precise mechanisms in disease progression is crucial for developing tailored therapeutic strategies to alleviate inflammatory and metabolic dysfunctions associated with these complex conditions.
ITGB2, a transmembrane glycoprotein pivotal for leukocyte adhesion and migration, facilitates immune cell recruitment and amplifies local inflammatory responses72. There are fewer studies on ITGB2 in PCOS, and we found that ITGB2 was significantly upregulated in PCOS patients and mouse models. We hypothesize that dysregulated ITGB2 expression correlates with intensified leukocyte infiltration and heightened inflammation, contributing to ovarian dysfunction and metabolic disturbances observed in PCOS patients. Similarly, in COVID-19, ITGB2-mediated leukocyte adhesion is crucial for orchestrating immune responses pivotal in disease progression73. ITGB2 promotes inflammatory cell recruitment to infection sites, exacerbating cytokine release and systemic inflammation. Elevated ITGB2 levels are associated with severe COVID-19 manifestations such as cytokine storms and acute respiratory distress syndrome, highlighting its role in modulating immune-mediated lung injury and vascular dysfunction74. Ongoing research into ITGB2’s molecular mechanisms and therapeutic implications is crucial for advancing precision medicine approaches aimed at mitigating inflammatory and metabolic dysfunctions associated with these complex conditions.
This study also revealed the interaction of hub genes with ovary-specific genes. GO and KEGG Pathway analyses showed significant enrichment in inflammatory pathways, ovarian follicle development, and insulin signaling pathways. According to our findings in the PPI network, TLR2 interacted with MYD88, TLR1, IRF3, and other key players in the inflammatory response signaling pathway75,76. S100A9 could potentially interact with S100A8, and its complex was believed to facilitate cyst migration in PCOS development53. S100A9 could increase the production of inflammatory cytokines and disturb the steroidogenesis of PCOS77. It has been reported that S100A8 and S100A9 have diagnostic biomarker values and can be used to identify COVID-19 patients admitted to intensive care units78. Hence, S100A8 and S100A9 appeared to have important roles in causing COVID-19, as well as immune responses.
In addition, we also found that EGFR proteins had direct or indirect interactions with S100A9, TLR2, ITGB, and ICAM1. As a critical factor in cell growth, differentiation, implantation, and decidualization, EGFR was vital for reproduction79. It is believed that the EGFR was responsible for facilitating proliferation and inhibiting apoptosis of granulosa cells by activating the MAPK/ERK signaling pathway and inducing transcription factor AP1 expression80. According to other studies, EGFR was expressed by cells of the lungs after SARS-CoV-2 infection. An elevated level of EGFR expression can further exacerbate pulmonary disease and cause fibrosis. Nimotuzumab can block the EGFR, which could be a novel treatment strategy for COVID-1981.
We have identified micro-RNA species has-miR-21-5p, has-miR-335-5p, has-miR-146a-5p, and has-miR-143-5p shared by the hub genes. These micro-RNAs have been linked to the development of PCOS82,83,84. Among them, has-miR-335-5p and has-miR-146a-5p were demonstrated to be involved in the DEGs-miRNA network regulation in COVID-1985. In particular, our previous study displayed that miR-335-5p may function as a mediator in the etiopathogenesis of PCOS and has the potential as both a novel diagnostic biomarker and therapeutic target for PCOS19. Establishing a hub gene-miRNA network might provide new insights into the pathogenesis of PCOS and COVID-19.
We also identified TFs closely associated with the hub genes. For instance, we identified STAT3, which participated in both TLR2 and S100A9 transcription. STAT3 has been shown to regulate the expression of TLR2 and S100A9. Given the roles of TLR2 and SA100A9 discussed above, we speculated that STAT3 might have the same function in PCOS and COVID-1986. Additionally, our previous study found that STAT3 could act directly on the miR-27a-3p promoter to induce granulosa cell apoptosis during the development of PCOS34.
The drugs and chemical compounds found in our analysis include resveratrol, lipopolysaccharides, methotrexate, nickel, tretinoin, Captopril, and Glucosamine. For instance, tretinoin has been stipulated to regulate steroid biosynthesis in human ovarian theca cells87. In addition, resveratrol was used to regulate inflammation and oxidative stress of granulosa cells in PCOS via targeting TLR2, which was consistent with our above finding of TLR2 as a hub gene88.
Conclusion
In the present study, we explored the mechanisms of common pathogenetic processes between SARS-CoV-2 infection and polycystic ovary syndrome based on an integrated bioinformatics approach. We mined seven common hub genes and validated their expression in clinical samples and mouse models. Six hub genes of S100A9, CD86, ICAM, THBD, MMP9, and ITGB2 exhibited a desirable performance in the diagnosis of COVID-19 and PCOS. Immune microenvironment analysis revealed that the hub genes were positively correlated with immune scores and significantly correlated with T cells, dendritic cells, B cells, NK cells, and eosinophils. This suggests that the inflammatory state of PCOS may exacerbate the immune response to SARS-CoV-2 infection. This study may provide a theoretical basis and innovative ideas for further mechanistic research and drug discovery of the comorbidity of two diseases.
Data availability
All data are derived from publicly available sources, and all links to the data are provided in the materials and methods.
Abbreviations
- PCOS:
-
Polycystic ovarian syndrome
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PPI:
-
Protein-protein interactions
- DEGs:
-
Differentially expressed genes
- ROC:
-
Receiver operating characteristic curve
- TF:
-
Transcription factor
- GSEA:
-
Gene set enrichment analysis
References
Xu, M. et al. Effects of dietary grape seed proanthocyanidin extract supplementation on meat quality, muscle fiber characteristics and antioxidant capacity of finishing pigs. Food Chem.367, 130781. https://doi.org/10.1016/j.foodchem.2021.130781 (2022).
Horvath, E. et al. Photocatalytic nanowires-based air filter: towards reusable protective masks. Adv. Funct. Mater.30, 2004615. https://doi.org/10.1002/adfm.202004615 (2020).
Williamson, E. J. et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature584, 430–436. https://doi.org/10.1038/s41586-020-2521-4 (2020).
Subramanian, A. et al. Increased COVID-19 infections in women with polycystic ovary syndrome: a population-based study. Eur. J. Endocrinol.184, 637–645. https://doi.org/10.1530/EJE-20-1163 (2021).
Takahashi, T. et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature588, 315–320. https://doi.org/10.1038/s41586-020-2700-3 (2020).
Jin, J. M. et al. Gender differences in patients with COVID-19: focus on severity and mortality. Front. Public Health8, 152. https://doi.org/10.3389/fpubh.2020.00152 (2020).
Peckham, H. et al. Male sex identified by global COVID-19 meta-analysis as a risk factor for death and ITU admission. Nat. Commun.11, 6317. https://doi.org/10.1038/s41467-020-19741-6 (2020).
Ma, L. et al. Evaluation of sex-related hormones and semen characteristics in reproductive-aged male COVID-19 patients. J. Med. Virol.93, 456–462. https://doi.org/10.1002/jmv.26259 (2021).
Suba, Z. Prevention and therapy of COVID-19 via exogenous estrogen treatment for both male and female patients. J. Pharm. Pharm. Sci.23, 75–85. https://doi.org/10.18433/jpps31069 (2020).
Stelzig, K. E. et al. Estrogen regulates the expression of SARS-CoV-2 receptor ACE2 in differentiated airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol.318, L1280–L1281. https://doi.org/10.1152/ajplung.00153.2020 (2020).
Lucas, J. M. et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 4, 1310–1325. https://doi.org/10.1158/2159-8290.CD-13-1010 (2014).
Wang, M., An, K., Huang, J., Mprah, R. & Ding, H. A novel model based on necroptosis to assess progression for polycystic ovary syndrome and identification of potential therapeutic drugs. Front. Endocrinol. (Lausanne). 14, 1193992. https://doi.org/10.3389/fendo.2023.1193992 (2023).
Rosenfield, R. L. & Ehrmann, D. A. The pathogenesis of polycystic ovary syndrome (PCOS): the hypothesis of PCOS as functional ovarian hyperandrogenism revisited. Endocr. Rev.37, 467–520. https://doi.org/10.1210/er.2015-1104 (2016).
Montopoli, M. et al. Androgen-deprivation therapies for prostate cancer and risk of infection by SARS-CoV-2: a population-based study (N = 4532). Ann. Oncol.31, 1040–1045. https://doi.org/10.1016/j.annonc.2020.04.479 (2020).
Fox, C. W. et al. Inflammatory stimuli trigger increased androgen production and shifts in Gene expression in Theca-interstitial cells. Endocrinology160, 2946–2958. https://doi.org/10.1210/en.2019-00588 (2019).
Kyrou, I. et al. Polycystic ovary syndrome (PCOS) and COVID-19: an overlooked female patient population at potentially higher risk during the COVID-19 pandemic. BMC Med.18, 220. https://doi.org/10.1186/s12916-020-01697-5 (2020).
Lopez-Martinez, C. et al. Transcriptomic clustering of critically ill COVID-19 patients. Eur. Respir. J.https://doi.org/10.1183/13993003.00592-2022 (2022).
Overmyer, K. A. et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst. 12, 23–40 e27. https://doi.org/10.1016/j.cels.2020.10.003 (2021).
Zhang, S. et al. Role and mechanism of mir-335-5p in the pathogenesis and treatment of polycystic ovary syndrome. Transl Res.https://doi.org/10.1016/j.trsl.2022.07.007 (2022).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics26, 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2010).
Chen, H. & Boutros, P. C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform.12, 35. https://doi.org/10.1186/1471-2105-12-35 (2011).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res.28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res.51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res.47, D607–D613. https://doi.org/10.1093/nar/gky1131 (2019).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res.13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Bader, G. D. & Hogue, C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform.4. https://doi.org/10.1186/1471-2105-4-2 (2003).
Chin, C. H. et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol.8 (Suppl 4). https://doi.org/10.1186/1752-0509-8-S4-S11 (2014).
Franz, M. et al. GeneMANIA update 2018. Nucleic Acids Res.46, W60–W64. https://doi.org/10.1093/nar/gky311 (2018).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A.102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics27, 1739–1740. https://doi.org/10.1093/bioinformatics/btr260 (2011).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol.37, 773–782. https://doi.org/10.1038/s41587-019-0114-2 (2019).
Xia, J., Gill, E. E. & Hancock, R. E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc.10, 823–844. https://doi.org/10.1038/nprot.2015.052 (2015).
Fornes, O. et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res.48, D87–D92. https://doi.org/10.1093/nar/gkz1001 (2020).
Wang, M. et al. Functional characterization of MicroRNA-27a-3p expression in human polycystic ovary syndrome. Endocrinology159, 297–309. https://doi.org/10.1210/en.2017-00219 (2018).
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform.12, 77. https://doi.org/10.1186/1471-2105-12-77 (2011).
Khateri, S., Mohammadi, H., Khateri, R. & Moradi, Y. The prevalence of underlying diseases and comorbidities in COVID-19 patients; an updated systematic review and meta-analysis. Arch. Acad. Emerg. Med.8, e72 (2020).
Barbaro, R. P. et al. Extracorporeal membrane oxygenation support in COVID-19: an international cohort study of the extracorporeal life support organization registry. Lancet396, 1071–1078. https://doi.org/10.1016/S0140-6736(20)32008-0 (2020).
Yunna, C., Mengru, H., Lei, W. & Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol.877, 173090. https://doi.org/10.1016/j.ejphar.2020.173090 (2020).
Rudnicka, E. et al. Chronic low grade inflammation in pathogenesis of PCOS. Int. J. Mol. Sci.22. https://doi.org/10.3390/ijms22073789 (2021).
Jiang, J. et al. Enhanced signaling through the TLR9 pathway is associated with resistance to HIV-1 infection in Chinese HIV-1-exposed seronegative individuals. Front. Immunol.11, 1050. https://doi.org/10.3389/fimmu.2020.01050 (2020).
Wu, H. M., Zhao, C. C., Xie, Q. M., Xu, J. & Fei, G. H. TLR2-melatonin feedback Loop regulates the activation of NLRP3 inflammasome in murine allergic airway inflammation. Front. Immunol.11, 172. https://doi.org/10.3389/fimmu.2020.00172 (2020).
Zheng, M. et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol.22, 829–838. https://doi.org/10.1038/s41590-021-00937-x (2021).
Khan, S. et al. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. Elife10. https://doi.org/10.7554/eLife.68563 (2021).
Gu, B. X. et al. Abnormal expression of TLRs may play a role in lower embryo quality of women with polycystic ovary syndrome. Syst. Biol. Reprod. Med.62, 353–358. https://doi.org/10.1080/19396368.2016.1187683 (2016).
Ernst, E. H. et al. Distinct expression patterns of TLR transcripts in human oocytes and granulosa cells from primordial and primary follicles. J. Reprod. Immunol.140, 103125. https://doi.org/10.1016/j.jri.2020.103125 (2020).
Yuan, B. et al. Resveratrol regulates the inflammation and oxidative stress of granulosa cells in PCOS via targeting TLR2. J. Bioenerg Biomembr.. https://doi.org/10.1007/s10863-022-09942-7 (2022).
Gonen, M. et al. Rapid and effective vitamin D supplementation may present better clinical outcomes in COVID-19 (SARS-CoV-2) patients by altering serum INOS1, IL1B, IFNg, Cathelicidin-LL37, and ICAM1. Nutrients13. https://doi.org/10.3390/nu13114047 (2021).
Rashad, N. M. et al. Intercellular adhesion molecule-1 expression and serum levels as markers of pre-clinical atherosclerosis in polycystic ovary syndrome. J. Ovarian Res.12, 97. https://doi.org/10.1186/s13048-019-0566-5 (2019).
Akcali, A. et al. Gingival inflammation and salivary or serum granulocyte-secreted enzymes in patients with polycystic ovary syndrome. J. Periodontol. 88, 1145–1152. https://doi.org/10.1902/jop.2017.170043 (2017).
Colicchia, M. et al. S100A8/A9 drives the formation of procoagulant platelets through GPIbalpha. Blood140, 2626–2643. https://doi.org/10.1182/blood.2021014966 (2022).
Fan, Z. P. et al. S100A9 activates the immunosuppressive switch through the PI3K/Akt pathway to maintain the immune suppression function of testicular macrophages. Front. Immunol.12, 743354. https://doi.org/10.3389/fimmu.2021.743354 (2021).
Ursino, G. et al. S100A9 exerts insulin-independent antidiabetic and anti-inflammatory effects. Sci. Adv.10, eadj4686. https://doi.org/10.1126/sciadv.adj4686 (2024).
Manibalan, S. et al. Protein network studies on PCOS biomarkers with s100A8, druggability assessment, and RNA aptamer designing to control its cyst migration effect. Front. Bioeng. Biotechnol.8, 328. https://doi.org/10.3389/fbioe.2020.00328 (2020).
Mellett, L. & Khader, S. A. S100A8/A9 in COVID-19 pathogenesis: impact on clinical outcomes. Cytokine Growth Factor Rev.63, 90–97. https://doi.org/10.1016/j.cytogfr.2021.10.004 (2022).
Qureshi, O. S. et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science332, 600–603. https://doi.org/10.1126/science.1202947 (2011).
Kennedy, A. et al. Differences in CD80 and CD86 transendocytosis reveal CD86 as a key target for CTLA-4 immune regulation. Nat. Immunol.23, 1365–1378. https://doi.org/10.1038/s41590-022-01289-w (2022).
Wang, F. et al. Discovery of microglia gonadotropin–releasing hormone receptor and its potential role in polycystic ovarian syndrome. Mol. Med. Rep.27. https://doi.org/10.3892/mmr.2023.12964 (2023).
Xie, D. et al. A toll-like receptor-activating, self-adjuvant glycan nanocarrier. Front. Chem.10, 864206. https://doi.org/10.3389/fchem.2022.864206 (2022).
Martin, T. M., Burke, S. J., Wasserfall, C. H. & Collier, J. J. Islet beta-cells and intercellular adhesion molecule-1 (ICAM-1): integrating immune responses that influence autoimmunity and graft rejection. Autoimmun. Rev.22, 103414. https://doi.org/10.1016/j.autrev.2023.103414 (2023).
Birnhuber, A. et al. Between inflammation and thrombosis: endothelial cells in COVID-19. Eur. Respir. J.58. https://doi.org/10.1183/13993003.00377-2021 (2021).
Smith-Norowitz, T. A., Loeffler, J., Norowitz, Y. M. & Kohlhoff, S. Intracellular adhesion molecule-1 (ICAM-1) levels in convalescent COVID-19 serum: a case report. Ann. Clin. Lab. Sci.51, 730–734 (2021).
Pan, C. C. et al. Antagonizing the irreversible thrombomodulin-initiated proteolytic signaling alleviates age-related liver fibrosis via senescent cell killing. Cell. Res.33, 516–532. https://doi.org/10.1038/s41422-023-00820-4 (2023).
Giri, H. et al. Thrombomodulin is essential for maintaining quiescence in vascular endothelial cells. Proc. Natl. Acad. Sci. U. S. A.118. https://doi.org/10.1073/pnas.2022248118 (2021).
Asteris, P. G. et al. Genetic prediction of ICU hospitalization and mortality in COVID-19 patients using artificial neural networks. J. Cell. Mol. Med.26, 1445–1455. https://doi.org/10.1111/jcmm.17098 (2022).
van Moorsel, M. V. A. et al. VhH anti-thrombomodulin clone 1 inhibits TAFI activation and enhances fibrinolysis in human whole blood under flow. J. Thromb. Haemost.20, 1213–1222. https://doi.org/10.1111/jth.15674 (2022).
Won, T. et al. Endothelial thrombomodulin downregulation caused by hypoxia contributes to severe infiltration and coagulopathy in COVID-19 patient lungs. EBioMedicine75, 103812. https://doi.org/10.1016/j.ebiom.2022.103812 (2022).
Zhang, H. et al. MMP9 protects against LPS-induced inflammation in osteoblasts. Innate Immun.26, 259–269. https://doi.org/10.1177/1753425919887236 (2020).
Ranjbaran, J. et al. Matrix metalloproteinases 2 and 9 and MMP9/NGAL complex activity in women with PCOS. Reproduction151, 305–311. https://doi.org/10.1530/REP-15-0340 (2016).
Wang, Y. et al. Pathway and network analyses identify growth factor signaling and MMP9 as potential mediators of mitochondrial dysfunction in severe COVID-19. Int. J. Mol. Sci.24. https://doi.org/10.3390/ijms24032524 (2023).
Gelzo, M. et al. Matrix metalloproteinases (MMP) 3 and 9 as biomarkers of severity in COVID-19 patients. Sci. Rep.12, 1212. https://doi.org/10.1038/s41598-021-04677-8 (2022).
Vaz de Paula, C. B. et al. COVID-19: immunohistochemical analysis of TGF-beta signaling pathways in Pulmonary Fibrosis. Int. J. Mol. Sci.23https://doi.org/10.3390/ijms23010168 (2021).
Zhang, X. et al. ITGB2-mediated metabolic switch in CAFs promotes OSCC proliferation by oxidation of NADH in mitochondrial oxidative phosphorylation system. Theranostics10, 12044–12059. https://doi.org/10.7150/thno.47901 (2020).
Siekacz, K. et al. Soluble ITGaM and ITGb2 integrin subunits are involved in Long-Term Pulmonary complications after COVID-19 infection. J. Clin. Med.12https://doi.org/10.3390/jcm12010342 (2023).
Cao, X. et al. Single-cell transcriptome landscape and antigen receptor dynamic during SARS-CoV-2 vaccination. Genes Dis.10, 1675–1686. https://doi.org/10.1016/j.gendis.2022.08.020 (2023).
Di Padova, F., Quesniaux, V. F. J. & Ryffel, B. MyD88 as a therapeutic target for inflammatory lung diseases. Expert Opin. Ther. Targets22, 401–408. https://doi.org/10.1080/14728222.2018.1464139 (2018).
IRF3. IRF7 require SIRT1 for liquid-liquid phase separation and transactivation of IFN-1. Nat. Immunol.23, 1144–1145. https://doi.org/10.1038/s41590-022-01270-7 (2022).
Li, H. et al. S100-A9 protein in exosomes derived from follicular fluid promotes inflammation via activation of NF-kappaB pathway in polycystic ovary syndrome. J. Cell. Mol. Med.24, 114–125. https://doi.org/10.1111/jcmm.14642 (2020).
Chen, L. et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol.17, 992–994. https://doi.org/10.1038/s41423-020-0492-x (2020).
Romano, R. & Bucci, C. Role of EGFR in the nervous system. Cells9. https://doi.org/10.3390/cells9081887 (2020).
Zheng, Q. et al. ANP promotes proliferation and inhibits apoptosis of ovarian granulosa cells by NPRA/PGRMC1/EGFR complex and improves ovary functions of PCOS rats. Cell. Death Dis.8, e3145. https://doi.org/10.1038/cddis.2017.494 (2017).
Londres, H. D. et al. Blocking EGFR with nimotuzumab: a novel strategy for COVID-19 treatment. Immunotherapy14, 521–530. https://doi.org/10.2217/imt-2022-0027 (2022).
Cao, M. et al. Adipose mesenchymal stem cell-derived exosomal microRNAs ameliorate polycystic ovary syndrome by protecting against metabolic disturbances. Biomaterials288, 121739. https://doi.org/10.1016/j.biomaterials.2022.121739 (2022).
Zhang, C. et al. MiRNAs expression profiling of rat ovaries displaying PCOS with insulin resistance. Arch. Gynecol. Obstet.302, 1205–1213. https://doi.org/10.1007/s00404-020-05730-z (2020).
Tian-Min, Y. et al. Combined transcriptomic and metabolomic analysis of women with polycystic ovary syndrome. Dis Mark. 4000424. https://doi.org/10.1155/2022/4000424 (2022).
Zhang, H. & Feng, T. Network-based data analysis reveals ion channel-related gene features in COVID-19: a bioinformatic approach. Biochem. Genet.https://doi.org/10.1007/s10528-022-10280-x (2022).
Feng, L. et al. Exercise Training protects Against Heart failure Via Expansion of myeloid-derived suppressor cells through regulating IL-10/STAT3/S100A9 pathway. Circ. Heart Fail.15, e008550. https://doi.org/10.1161/CIRCHEARTFAILURE.121.008550 (2022).
Wickenheisser, J. K. et al. Retinoids and retinol differentially regulate steroid biosynthesis in ovarian theca cells isolated from normal cycling women and women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab.90, 4858–4865. https://doi.org/10.1210/jc.2005-0330 (2005).
Krause, K. L., Volz, K. W. & Lipscomb, W. N. 2.5 a structure of aspartate carbamoyltransferase complexed with the bisubstrate analog N-(phosphonacetyl)-L-aspartate. J. Mol. Biol.193, 527–553. https://doi.org/10.1016/0022-2836(87)90265-8 (1987).
Acknowledgements
We thank the depositors of the GSE34526, GSE10946, GSE147507, GSE157103, GSE54248, and GSE164805 datasets for their contribution. We thank Dr. Richard Mprah for his help in revising and polishing the language.
Funding
This research was financially supported by the National Natural Science Foundation of China (Grant No. 82401923) and the Natural Science Research of the Jiangsu Higher Education Institutions of China (Grant No. 22KJB180007) and the Outstanding Talent Research Funding of Xuzhou Medical University (Grant No. RC20552029).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study’s conception and design. Hai Bai and Shanshan Zhang performed data analysis, qPCR testing, and manuscript writing. Hai Bai performed material preparation and data collection. Cui Li and Mingming Wang performed manuscript revising. Jing Huang and Kangyang Diao contributed to the revision of the paper’s images and content. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bai, H., Zhang, S., Huang, J. et al. Unraveling the pathogenic interplay between SARS-CoV-2 and polycystic ovary syndrome using bioinformatics and experimental validation. Sci Rep 14, 22934 (2024). https://doi.org/10.1038/s41598-024-74347-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-74347-y
