
Abstract
The novel coronavirus disease 2019 (COVID-19) is a global epidemic caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). SARS-CoV-2 has a similar structure to severe acute respiratory syndrome coronavirus-1(SARS-CoV-1). The S protein on the surface of the virus is cleaved by host proprotein convertases (PCs) to expose the active N-terminal S1 extracellular domain. Its receptors are angiotensin-converting enzyme 2 (ACE2), and the C-terminal S2 membrane anchoring protein is responsible for translocating the virus into the cell. Among patients with COVID-19, there is a higher prevalence of cardiovascular disease, and more than 7% of patients have suffered myocardial damage due to the infection, but the internal mechanism is still poorly understood. There is currently no specific and effective targeted treatment. Reduction of the patient’s morbidity and mortality is an urgent problem that needs to be solved clinically. By exploring the theoretical analysis of PCs and ACE2 in COVID-19 cardiovascular susceptibility, some insights on how to prevent and alleviate adverse cardiovascular prognosis have been provided in this study.
Similar content being viewed by others

Introduction
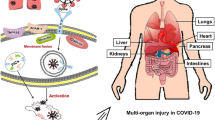
The epidemics caused by coronavirus before COVID-19, acute respiratory syndrome (SARS) [1, 2], and Middle East respiratory syndrome (MERS) [3, 4] outbreaks were reported to be related to cardiovascular disease. Current clinical reports indicate that SARS-CoV-2 is associated with significant morbidity of cardiovascular diseases and complications, such as hypertension (HTN), myocarditis, acute myocardial infarction, and increased heart failure [5, 6]. Researchers have extensively studied the pathophysiology of SARS-CoV-2 infection and believe that the ACE2 receptor acts as a high-affinity receptor and co-transporter of the virus into the cell [7, 8]. The widespread expression of ACE2 in the myocardium and vascular endothelial cells might be due to the presence of SARS-CoV-2, which could cause direct cardiovascular damage and lead to adverse consequences [6, 9].
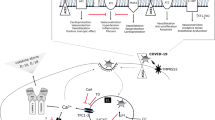
The S protein plays a vital role in the process of SARS-CoV-2 infecting the host and translocating into the cell [7, 10]. Coronavirus S protein is a granule-shaped structural protein with a length of about 1200 aa, which helps the virus to bind to cell receptors and participate in mediating viral infection and pathogenesis [11, 12]. During viral infection, the host cell PCs cleave the S1/S2 cleavage site of the trimer S protein dividing it into an N-terminal S1 extracellular domain that recognizes the relevant cell surface receptor and a C terminal S2 membrane anchor protein that is involved in translocation of the virus into the cells [13, 14]. SARS-Cov-S2 and SARS-CoV-S1 proteins contain a conserved receptor-binding domain (RBD), which can recognize ACE2 [15]. Therefore, PCs family, especially furin, can be considered the key medium that mediates the maturation of S protein processing and recognition of membrane proteins. Furin can also be considered a critical molecule that makes SARS-Cov-S2 cause adverse cardiovascular events through the ACE2 receptor. This speculation is also supported by the occurrence of a high level of furin in the peripheral blood of heart failure patients.
Based on these theoretical assumptions, it can be concluded that the S protein/host furin/ACE2 signal axis exists in the pathological process of SARS-Cov-S2 infection and mediates the occurrence of adverse cardiovascular prognosis events. Numerous studies discussed ACE2 before, and therefore this article has focused on the role of furin in this pathological process. Furthermore, a unique furin-like cleavage site exists in the S protein of SARS-Cov-S2 [16]; thus, the theoretical advantage inferred from this cleavage site in disease infection models can be deduced to prevent and combat COVID-19 caused by SARS-CoV-2.
COVID-19 and Cardiovascular Complications
COVID-19 is mainly presented with respiratory symptoms. Patients with severe infections usually present with acute respiratory distress syndrome (ARDS) and septic shock. The cardiovascular system involvement in this infection is also a significant feature. Cardiovascular disease (CVD) is a common comorbidity of COVID-19, SARS, and MERS patients. In SARS, the prevalence of diabetes mellitus (DM) and CVD is 11% and 8%, respectively, and their occurrence increases the risk of death [17]. About 50% of MERS cases are combined with DM and HTN, and about 30% are combined with CVD [18]. COVID-19 also has cardiovascular comorbidities, especially in cases with more severe conditions. By April 2020, the “Analysis of New Coronavirus Pneumonia Epidemiological Characteristics,” published by the Chinese Center for Disease Control and Prevention, indicated that hypertension was the most common underlying disease among the patients. There were 2683 patients with hypertension, accounting for 12.8% of the total number of patients, 39.7% of the dead patients had hypertension, 4.2% of patients and 22.7% of patients with cardiovascular disease [19], and more than 20% of severe patients had myocardial damage [20]. Besides, some patients showed symptoms of palpitations. In a study of 137 patients, 7.3% of patients had palpitations as the initial symptom [21]. Finally, a recent meta-analysis of eight studies from China, including a total of 46,248 infected patients, showed that the most common comorbidities were HTN (17 ± 7%, 95% CI 14–22%), DM (8 ± 6%, 95% CI 6–11%), and cardiovascular disease (5 ± 4%, 95% CI 4–7%) [22]. However, the mechanism of these associations is currently unclear. The current hypotheses include the common risk factors of CVD and COVID-19 (such as advanced age and low immunity), ACE2, and furin levels.
Proprotein Convertases and S Protein
The processing and activation of the coronavirus S protein are critical to the infectivity of the virus. Therefore, relevant enzymes constitute potential targets for antiviral intervention. The proprotein convertase family (PCs; gene name PCSKs) is composed of nine serine-secreting proteases and is widely involved in regulating various biological processes in normal and disease states. The PCs can perform multiple activation functions and are involved in many key cellular pathways. The PCs cleave precursor proteins at specific single or paired basic amino acids (aa), turning them precursor proteins into an active state. In many PCs, the processed substrate is the infectious virus cell surface glycoproteins [23]. The relationship between PCs (especially furin) and viral infection was confirmed based on their role in the processing of many essential cell surface proteins [24]. For human coronavirus infections, the S proteins of HcoV-OC43, MERS-CoV, and SARS-CoV show the catalytic motif of PCs: Lys/Arg-Xn-Lys/Arg↓ (where Xn is other amino acids except cysteine). Of amino acids, n can be 0, 2, 4, or 6↓ [25]. This further confirms the role of PCs in the mechanism of coronavirus infection.
The S protein acts as a key medium for SARS⁃CoV⁃2 to invade the host and cause cell damage. After entering the body, the host cell PCs cleave the trimer S protein at the S1/S2 cleavage site, dividing it into an N-terminal extracellular domain of S1, which recognizes related cell surface receptors, and a C-terminal S2 membrane anchoring protein, which is involved in the entry of the virus into the cells. SARS-Cov-S2, like the S protein of SARS-CoV-S1, contains a conserved RBD that recognizes ACE2 [26, 27]. Therefore, the PCs family, especially furin, can be regarded as a key medium that mediates the processing and maturation of viral spike protein and recognizes membrane proteins and plays an important role in the treatment process of viral infection [27, 28]. The biological process of Furin processing and activation of coronavirus S protein to expose the reactive domain also explains partially the phenomenon of COVID-19 with severe cardiovascular damage. Furthermore, heart failure, accompanied by a marked increase in the level of furin in the peripheral blood in the course of COVID-19 in patients [29], makes this conjecture more credible. Therefore, inhibition of such processing enzymes could represent a potential antiviral strategy.
Interestingly, members of the PCs family often cause functional redundancy or complementation due to the high structural similarity [30]. Among the PCs secreted by the heart, PCSK6 is the most abundant [31] and is more than 70% structurally similar to furin. PCSK6 functions as a critical protease for processing corin and is involved in the regulation of enkephalin blood pressure [32]. It has also been shown to be involved in the pathological process of atherosclerosis and myocardial fibrosis after myocardial infarction [33]. Therefore, furin is likely to be a functional complementarity or redundancy of PCSK6. Blocking the activity of these enzymes can reduce viral infections and cardiovascular damage and is beneficial for the treatment of cardiovascular diseases. Besides, to limit viral infection, host cells infected by the virus can cause an interferon response, which autonomously inhibits the enzyme activity of furin [34].
These previous findings have indicated that furin inhibitors could help in suppressing the spread of SARS-CoV-2. Moreover, based on the role of furin protein in cardiovascular disease, its inhibitors might also improve cardiovascular prognosis and reduce critical deaths. Therefore, furin could be a promising target for the development of new treatments.
Pathogens or their toxins require processing by host PCs to enter host cells and cause disease. Conversely, inhibiting PCs may protect host cells from furin-dependent pathogens. The correctness of the concept of furin-dependent infection has been supported by many basic studies [35,36,37]. Various methods have been proposed that inhibit furin activity hence limiting viral and bacterial infections and tumor growth. The variant of natural serine protease inhibitor α-1 antitrypsin affects the replication of the HIV-1 virus by interfering with the differentiation and maturation of gp160 to gp120 and gp41 [38]. Also, the clarification of the crystal structure of furin promotes the design of 2,5-dideoxystreptamine derivative inhibitors [39, 40]. Nevertheless, because furin is involved in many cellular processes, it is important to avoid this systemic inhibition as it can cause some toxicity. Therefore, rapid screening of small molecule inhibitors or other more effective oral active inhibitors, such as Andrographis paniculata [41], should be conducted to evaluate its antiviral effect on 2019-nCoV.
The abnormal expression or activity of furin can cause a variety of diseases that include infectious (viral or bacterial infections) and non-infectious diseases, metabolic diseases, and even cancer [34]. Furin has also been shown to be involved in S protein cleavage and SARS-CoV-2 pathogenicity [34]. Although furin has been regarded as a potential therapeutic target for infectious diseases, the use of host protease inhibitors (especially inhibitors against furin) as a treatment strategy for COVID-19 seems to be premature. Therefore, its lysis role and pathogenesis in SARS-CoV-2 need further clarification.
S Protein and ACE2
For coronavirus to enter the host target cell, it needs to complete two key steps. It first binds to the cell surface by attaching to the host cell receptor and then fuses its envelope to the cell membrane for the viral genome to be released into the cytoplasm of the host cell to achieve viral replication. Both of these steps are controlled by the S envelope protein [42].
S protein is a structural protein of about 1200 aa in length that constitutes the corona shape of coronavirus particles. It binds to the cell receptors and participates in mediating viral infection and pathogenesis [43]. However, in the process of infection, the S protein plays a direct damaging role by recognizing and binding to the ACE2 receptor and invading the host cell [10]. Studies have shown that the affinity of the S-spike protein of SARS⁃CoV⁃2 with ACE2 is 10 to 20 times higher than that of SARS⁃CoV-1 [44]. Based on the high expression of ACE2 in cardiomyocytes, it is theoretically speculated that many new coronaviruses could directly bind to ACE2 and directly damage cardiomyocytes.
On the other hand, S protein depletes in binding to ACE2, leading to myocardial damage mediated by ACE/ACE2 imbalance in vivo [45]. ACE2 is a homolog of ACE, but their functions are entirely different [46]. ACE-mediated endocrine regulation causes vasoconstriction and increased blood pressure through the ACE⁃AngII (Angiotensin II)⁃AT1 axis [47]. The ACE2⁃Ang 1⁃7⁃Mas axis mediated by ACE2 can antagonize the above effects [48]. AngII, as an inflammatory factor regulatory protein, plays an essential regulatory role in mediating myocardial injury, and ACE2 has a protective effect in organs such as the heart and kidney [45]. It is theoretically speculated that SARS⁃CoV⁃2 reduces the expression of ACE2 after infecting cardiomyocytes through S protein, causing an increase in AngII level, which results in cardiomyocyte damage and apoptosis.
Based on these theories, it is assumed that the ACE2 receptor plays an important role in this pathological process, and therefore by directly acting or by discontinuing ACEi/ARB, ACE2 has become a candidate treatment strategy [49]. The theoretical advantages of discontinuing ACEi/ARB from results observed in clinical cohort studies are inconsistent [50, 51]. Drug analysis in hypertensive-positive patients found no association between any single drug category and the increased likelihood of positive tests. At the same time, no drug is associated with a significant increase in the risk of serious diseases. The reduction of mortality caused by the use of ACEi/ARB has been adequately studied. The current beneficial effects on patients with diabetes, chronic kidney disease, and proteinuria or proteinuria exceed the theoretical risk. In patients with chronic heart failure, the beneficial effects of ACEi/ARB outweigh the theoretical risks. Currently, COVID-19 has reached a pandemic level and affect more patients with cardiovascular comorbidities, and the ongoing randomized clinical trials to investigate whether hospitalized COVID-19 patients should continue to use ACEi/ARB, will shed more light on the accuracy of the existing hypothesis (NCT04351581). Out of caution, there is currently no plan for discontinuation of ACEi/ARB in COVID-19 patients with heart failure, hypertension, or ischemic heart disease.
Faced with the dilemma of whether to discontinue ACEi/ARB, the clinical strategy of direct injection of ACE2 seems more promising. The bacterial-derived ACE2-like enzyme B38-CAP and human recombinant soluble angiotensin-converting enzyme 2 (rhACE2) have the same mechanism of action. The use of B38-CAP and rhACE2 can eliminate the membrane-anchoring domain to inhibit angiotensin II induction, high blood pressure, myocardial hypertrophy, and fibrosis, which in turn shows beneficial effects in disease models such as heart failure, acute lung injury, and diabetic nephropathy. The rhACE2 is currently being tested clinically to treat patients with ARDS and COVID-19 infection [52, 53]. On the other hand, hrsACE2 or B38-CAP can prevent SARS-CoV-2 from entering the cell and induce the virus to attach to the exogenous ACE2 copy instead of the host cell. This destroys the ability of the virus to infect the cell to the same extent leading to reduced viral replication in the lungs and other organs. Furthermore, the role of hrsACE2 or B38-CAP in inhibiting COVID-19 could be achieved by down-regulating the host ACE2 since it is the right receptor of SARS-CoV-2. Therefore, hrsACE2 or B38-CAP is expected to treat SARS-CoV-2 infection, and some of the ongoing clinical trials will further corroborate these clinical advantages (NCT04375046, NCT04382950).
Conclusion
At present, there is still a lack of in-depth research on the complete pathophysiological process of COVID-19 and cardiovascular disease. However, through clinical manifestations and theoretical assumptions, it is speculated that SARS⁃CoV⁃2 may affect the cardiovascular system through various mechanisms. Here, the S protein/Furin/ACE2 signal axis provides a further potential explanation for the susceptibility of the heart to COVID-19. The sensitivity of SARS-CoV-2 infection may be due to the simultaneous upregulation of ACE2 and furin in the diseased myocardium, and RAAS in this population. The lack of a specific antiviral drug means that the infected people cannot be treated and poses difficulties in controlling the spread of the virus. We mainly rely on isolation measures to prevent the spread of COVID-19. Although clinical trials of antiviral drug candidates (HIV protease inhibitors lopinavir and ritonavir) were conducted early in the outbreak, the test results were unfortunately disappointing. Chloroquine and hydroxychloroquine are not beneficial and increase the risk of arrhythmia or even death. It is still necessary to continue to test and apply the currently available and future therapies of COVID-19. Therefore, in patients with SARS-CoV-2 infection, transient regulation of the viral binding sites on ACE2 or furin through immunological or pharmacological methods may constitute a new therapeutic strategy to deal with this unprecedented and powerful virus threat.
Availability of Data and Material
All available information is contained within the present manuscript.
Abbreviations
- COVID-19:
-
novel coronavirus disease
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus-2
- SARS-CoV-1:
-
Severe acute respiratory syndrome coronavirus-1
- PCs:
-
Proprotein convertases
- ACE2:
-
Angiotensin converting enzyme 2
- SARS:
-
Severe acute respiratory syndrome coronavirus
- MERS:
-
Middle East respiratory syndrome
- HTN:
-
Hypertension
- RBD:
-
Receptor-binding domain
- ARDS:
-
Acute respiratory distress syndrome
- CVD:
-
Cardiovascular disease
- DM:
-
Diabetes mellitus
- AngII:
-
Angiotensin II
References
Peiris JS, Chu CM, Cheng VC, Chan KS, Hung IF, Poon LL, et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361(9371):1767–72. https://doi.org/10.1016/s0140-6736(03)13412-5.
Zhong NS, Zheng BJ, Li YM, Poon LLM, Xie ZH, Chan KH, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet. 2003;362(9393):1353–8. https://doi.org/10.1016/s0140-6736(03)14630-2.
Mizumoto K, Saitoh M, Chowell G, Miyamatsu Y, Nishiura H. Estimating the risk of Middle East respiratory syndrome (MERS) death during the course of the outbreak in the Republic of Korea, 2015. Int J Infect Dis. 2015;39:7–9. https://doi.org/10.1016/j.ijid.2015.08.005.
Matsuyama R, Nishiura H, Kutsuna S, Hayakawa K, Ohmagari N. Clinical determinants of the severity of Middle East respiratory syndrome (MERS): a systematic review and meta-analysis. BMC Public Health. 2016;16(1):1203. https://doi.org/10.1186/s12889-016-3881-4.
Fried JA, Ramasubbu K, Bhatt R, Topkara VK, Clerkin KJ, Horn E, et al. The Variety of Cardiovascular Presentations of COVID-19. Circulation. 2020;141:1930–6. https://doi.org/10.1161/circulationaha.120.047164.
Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, et al. COVID-19 and cardiovascular disease. Circulation. 2020;141(20):1648–55. https://doi.org/10.1161/circulationaha.120.046941.
Walls AC, Park YJ, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.e6. https://doi.org/10.1016/j.cell.2020.02.058.
Yan R, Zhang Y, Li Y, et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–8. https://doi.org/10.1126/science.abb2762.
Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;17(5):259–60. https://doi.org/10.1038/s41569-020-0360-5.
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–20. https://doi.org/10.1038/s41586-020-2180-5.
Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11(1):1620. https://doi.org/10.1038/s41467-020-15562-9.
Watanabe Y, Allen JD, Wrapp D, McLellan JS, Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science. 2020:eabb9983. https://doi.org/10.1126/science.abb9983.
Liu PP, Blet A, Smyth D, Li H. The science underlying COVID-19: implications for the cardiovascular system. Circulation. 2020;142:68–78. https://doi.org/10.1161/circulationaha.120.047549.
He WT, Ji X, He W, Dellicour S, Wang S, Li G, et al. Genomic epidemiology, evolution, and transmission dynamics of porcine deltacoronavirus. Mol Biol Evol. 2020. https://doi.org/10.1093/molbev/msaa117.
Yuan M, Wu NC, Zhu X, et al. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science. 2020;368(6491):630–3. https://doi.org/10.1126/science.abb7269.
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res. 2020;176:104742. https://doi.org/10.1016/j.antiviral.2020.104742.
Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med. 2003;348(20):1986–94. https://doi.org/10.1056/NEJMoa030685.
Atabani SF, Wilson S, Overton-Lewis C, Workman J, Kidd IM, Petersen E, et al. Active screening and surveillance in the United Kingdom for Middle East respiratory syndrome coronavirus in returning travellers and pilgrims from the Middle East: a prospective descriptive study for the period 2013-2015. Int J Infect Dis. 2016;47:10–4. https://doi.org/10.1016/j.ijid.2016.04.016.
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–20. https://doi.org/10.1056/NEJMoa2002032.
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. Jama. 2020;323(11):1061–9. https://doi.org/10.1001/jama.2020.1585.
Liu K, Fang YY, Deng Y, et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin Med J. 2020;133(9):1025–31. https://doi.org/10.1097/cm9.0000000000000744.
Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, et al. Prevalence of comorbidities and its effects in coronavirus disease 2019 patients: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91–5. https://doi.org/10.1016/j.ijid.2020.03.017.
Nakayama K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem J. 1997;327(Pt 3):625–35. https://doi.org/10.1042/bj3270625.
Millet JK, Whittaker GR. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci U S A. 2014;111(42):15214–9. https://doi.org/10.1073/pnas.1407087111.
Liu J, Zheng X, Tong Q, et al. Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS-CoV, MERS-CoV, and 2019-nCoV. J Med Virol. 2020;92(5):491–4. https://doi.org/10.1002/jmv.25709.
Xia S, Liu M, Wang C, Xu W, Lan Q, Feng S, et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30(4):343–55. https://doi.org/10.1038/s41422-020-0305-x.
Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A. 2020;117(21):11727–34. https://doi.org/10.1073/pnas.2003138117.
Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. 2020;78:779–784.e5. https://doi.org/10.1016/j.molcel.2020.04.022.
Semenov AG, Tamm NN, Seferian KR, Postnikov AB, Karpova NS, Serebryanaya DV, et al. Processing of pro-B-type natriuretic peptide: furin and corin as candidate convertases. Clin Chem. 2010;56(7):1166–76. https://doi.org/10.1373/clinchem.2010.143883.
Seidah NG, Sadr MS, Chrétien M, Mbikay M. The multifaceted proprotein convertases: their unique, redundant, complementary, and opposite functions. J Biol Chem. 2013;288(30):21473–81. https://doi.org/10.1074/jbc.R113.481549.
Kuhn TC, Knobel J, Burkert-Rettenmaier S, Li X, Meyer IS, Jungmann A, et al. Secretome analysis of cardiomyocytes identifies PCSK6 as a novel player in cardiac remodeling after myocardial infarction. Circulation. 2020;141:1628–44. https://doi.org/10.1161/circulationaha.119.044914.
Chen S, Cao P, Dong N, Peng J, Zhang C, Wang H, et al. PCSK6-mediated corin activation is essential for normal blood pressure. Nat Med. 2015;21(9):1048–53. https://doi.org/10.1038/nm.3920.
Rykaczewska U, Suur BE, Röhl S, Razuvaev A, Lengquist M, Sabater-Lleal M, et al. PCSK6 is a key protease in the control of smooth muscle cell function in vascular remodeling. Circ Res. 2020;126(5):571–85. https://doi.org/10.1161/circresaha.119.316063.
Braun E, Sauter D. Furin-mediated protein processing in infectious diseases and cancer. Clin Transl Immunology. 2019;8(8):e1073. https://doi.org/10.1002/cti2.1073.
Shiryaev SA, Remacle AG, Ratnikov BI, Nelson NA, Savinov AY, Wei G, et al. Targeting host cell furin proprotein convertases as a therapeutic strategy against bacterial toxins and viral pathogens. J Biol Chem. 2007;282(29):20847–53. https://doi.org/10.1074/jbc.M703847200.
Gagnon H, Beauchemin S, Kwiatkowska A, Couture F, D’Anjou F, Levesque C, et al. Optimization of furin inhibitors to protect against the activation of influenza hemagglutinin H5 and Shiga toxin. J Med Chem. 2014;57(1):29–41. https://doi.org/10.1021/jm400633d.
Seidah NG, Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov. 2012;11(5):367–83. https://doi.org/10.1038/nrd3699.
Bahbouhi B, Bendjennat M, Chiva C. Effect of alpha-1 antitrypsin Portland variant (α1-PDX) on HIV-1 replication. Biochem J. 2000;352(1):91–8. https://doi.org/10.1042/bj3520091.
Jiao GS, Cregar L, Wang J, Millis SZ, Tang C, O'Malley S, et al. Synthetic small molecule furin inhibitors derived from 2,5-dideoxystreptamine. Proc Natl Acad Sci U S A. 2006;103(52):19707–12. https://doi.org/10.1073/pnas.0606555104.
Dahms SO, Jiao GS, Than ME. Structural studies revealed active site distortions of human furin by a small molecule inhibitor. ACS Chem Biol. 2017;12(9):2474. https://doi.org/10.1021/acschembio.7b00633.
Jarukamjorn K, Nemoto N. Pharmacological aspects of Andrographis paniculata on health and its major diterpenoid constituent andrographolide. J Health Sci. 2008;54:370–81. https://doi.org/10.1248/jhs.54.370.
Snijder EJ, Gallagher T, Perlman S. Nidoviruses || Nidovirus Entry into Cells. 2008. https://doi.org/10.1128/9781555815790:157-178.
Chen Y, Qiu F. Spike protein in the detection and treatment of novel coronavirus. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2020;37(2):246–50. https://doi.org/10.7507/1001-5515.202002050.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–3. https://doi.org/10.1126/science.abb2507.
Brojakowska A, Narula J, Shimony R, Bander J. Clinical implications of SARS-Cov2 interaction with renin angiotensin system. J Am Coll Cardiol. 2020;75:3085–95. https://doi.org/10.1016/j.jacc.2020.04.028.
Zisman LS. ACE and ACE2: a tale of two enzymes. Eur Heart J. 2005;26(4):322–4. https://doi.org/10.1093/eurheartj/ehi043.
Arendse LB, Danser AHJ, Poglitsch M, Touyz RM, Burnett JC Jr, Llorens-Cortes C, et al. Novel therapeutic approaches targeting the renin-angiotensin system and associated peptides in hypertension and heart failure. Pharmacol Rev. 2019;71(4):539–70. https://doi.org/10.1124/pr.118.017129.
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/angiotensin-(1-7)/MAS Axis of the renin-angiotensin system: focus on angiotensin-(1-7). Physiol Rev. 2018;98(1):505–53. https://doi.org/10.1152/physrev.00023.2016.
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586–90. https://doi.org/10.1007/s00134-020-05985-9.
Reynolds HR, Adhikari S, Pulgarin C, Troxel AB, Iturrate E, Johnson SB, et al. Renin-angiotensin-aldosterone system inhibitors and risk of Covid-19. N Engl J Med. 2020;382:2441–8. https://doi.org/10.1056/NEJMoa2008975.
Mehta N, Kalra A, Nowacki AS, Anjewierden S, Han Z, Bhat P, et al. Association of use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1855.
Zhang X, Li S, Niu S. ACE2 and COVID-19 and the resulting ARDS. Postgrad Med J. 2020;96:403–7. https://doi.org/10.1136/postgradmedj-2020-137935.
Monteil V, Kwon H, Prado P, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181(4):905–913.e7. https://doi.org/10.1016/j.cell.2020.04.004.
Funding
The work was supported partly by the Shenzhen Science and Technology Research and Development Fund (NO JCY20180302173849459).
Author information
Authors and Affiliations
Contributions
YM and LQ were contributors writing the manuscript. All authors have read and approved the submitted manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Covid-19
Rights and permissions
About this article

Cite this article
Ming, Y., Qiang, L. Involvement of Spike Protein, Furin, and ACE2 in SARS-CoV-2-Related Cardiovascular Complications. SN Compr. Clin. Med. 2, 1103–1108 (2020). https://doi.org/10.1007/s42399-020-00400-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42399-020-00400-2