
Abstract
Four distinct species of human coronaviruses (HCoVs) circulate in humans. Despite the recent attention due to SARS-CoV-2, a comprehensive understanding of the molecular epidemiology and genomic evolution of HCoVs remains unclear. Here, we employed primary differentiated human nasal epithelial cells for the successful isolation and genome sequencing of HCoVs derived from two retrospective cohorts in Singapore and Tanzania. Phylodynamic inference shows that HCoV-229E and HCoV-OC43 were subject to stronger genetic drift and reduced purifying selection from the early 2000s onwards, primarily targeting spike Domain A and B. This resulted in increased lineage diversification, coinciding with a higher effective reproductive number (Re>1.0). However, HCoV-NL63 and HCoV-HKU1 experienced weaker genetic drift and selective pressure with prolonged regional persistence. Our findings suggest that HCoV-229E and HCoV-OC43 viruses are adept at generating new variants and achieving widespread intercontinental dissemination driven by continuous genetic drift, recombination, and complex migration patterns.
Similar content being viewed by others

Introduction
While SARS-CoV-2 remains a significant public health challenge, there are four species of human coronaviruses (HCoVs) that have persisted in humans since 1960, causing significant illness and mortality worldwide. Two species, HCoV-229E and HCoV-NL63, belong to the Alphacoronavirus genus, and the two other species, HCoV-OC43 and HCoV-HKU1, belong to the Betacoronavirus genus. All four species are zoonotic and it has been postulated that HCoV-229E1 and HCoV-NL632 may have originated from bats, whereas HCoV-OC43 and HCoV-HKU1 may have a rodent origin3. Human coronaviruses are associated with the common cold and typically cause mild illness, but they can lead to severe illness such as pneumonia and death, particularly among vulnerable groups, including young infants, children and the elderly4,5. Human seasonal coronaviruses are detected seasonally in temperate countries, with infections peaking between October and April6. The predominant species may vary between epidemics, with typically at least one alphacoronavirus and betacoronavirus in co-circulation. Fluctuations in prevalence and reinfection capability of HCoVs is likely due to waning immunity and antigenic drift7,8.
The four HCoVs species have genomes ranging between 27 and 30 kb in length. The first two-thirds of the genome is ORF1ab that encodes for 16 non-structural proteins. The final one-third comprises structural proteins including the spike, envelope (E), membrane (M) and nucleocapsid (N). Accessory proteins, including both non-structural and structural, are interspersed and vary in location across species9. HCoV-OC43 and HCoV-HKU1 carry an additional structural protein, hemagglutinin-esterase (HE), that is unique to subgenus Embecovirus and which acts as a receptor-destroying enzyme that plays a role in virus infection10. The spike protein is central to virus entry and the four HCoV species differ in their receptor binding preferences: HCoV-229E binds to human aminopeptidase N receptor11, HCoV-NL63 binds to the angiotensin-converting enzyme 2 (ACE2) receptor12, HCoV-OC43 binds to 9-O-acetylated sialic acid13, and HCoV-HKU1 was recently shown to bind to transmembrane protease serine 2 (TMPRSS2)14. Coronaviruses may undergo recombination events during coinfection via a model known as discontinuous transcription15. Briefly, the RNA polymerase can dissociate and switch templates during RNA transcription, resulting in a recombinant virus that contains regions from two parental viruses. Recombination plays a significant role in driving coronavirus evolution and is known to occur across all coronaviruses. Novel recombinants can arise that are capable of replacing circulating strains and leading to alteration of phenotype16. This is evident by the Middle-East Respiratory Syndrome coronavirus (MERS-CoV) that has acquired a receptor binding domain through recombination, enabling the utilization of the human receptor dipeptidyl peptidase 4 for cell host entry17.
The coronavirus infectious disease 2019 (COVID-19) pandemic has led to the unprecedented sequencing of more than 16 million complete genomes of SARS-CoV-2, while less than five-hundred genomes are publicly available for all four HCoVs combined over the last 60 years (Supplementary Fig. 1). Despite the discovery of HCoVs in the early 1960s, the scarcity of surveillance and challenges in virus isolation have hindered research on HCoV diversity and transmission18. The first complete genome of HCoV-229E from a clinical isolate was only generated in 201219. Nonetheless, COVID-19 has provided incentive to better understand the adaptive evolution of HCoVs. Recent studies have shown that positive selection is acting on the receptor binding domain of the spike, ORF5, and envelope proteins20,21. In particular, antigenic drift is more apparent in HCoV-229E and HCoV-OC43 viruses8,22.
Human coronaviruses had stagnant genetic diversity during their early emergence, however, variations in epidemic patterns were observed in the past decade. While these viruses may occur year-round, particularly in tropical countries, the underlying processes driving changes in epidemic activities are not fully understood. Here, we generated new complete genomes of HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1 from patient samples collected in Singapore and Tanzania to analyse with publicly available sequences. We employed phylogenetic inference to reconstruct the evolutionary history of each HCoV species since their introduction into humans. We show significant lineage expansion of HCoV-229E and HCoV-OC43, accompanied by several key non-synonymous substitutions in the receptor binding domain (RBD) of the spike protein and comparatively stronger selective pressure. This contrasts with HCoV-NL63 and HCoV-HKU1 which are subject to weaker selection and exhibit slower drift. Additionally, we integrated population dynamics and phylogeographic methods to infer fluctuations over time in population patterns, epidemic growth, and spatial migration pathways of these co-circulating coronaviruses. A comprehensive understanding these factors is imperative for the development of effective strategies to control and mitigate future epidemics of these viruses.
Results
Genomic recombination landscape across human coronaviruses
Recent lineage diversification of HCoV-229E and HCoV-OC43 viruses. Between 2009 and 2018, we obtained more than 3042 nasal swab samples from two cohorts in Singapore and Tanzania. In total, we generated 31 complete genomes from the four HCoVs (Table 1), including HCoV-229E (n = 8), HCoV-OC43 (n = 11), HCoV-NL63 (n = 8), and HCoV-HKU1 (n = 4), that were analysed alongside all available global sequences to understand the changes in relative genetic diversity and population dynamics of HCoV species.
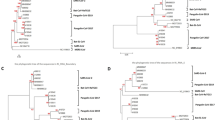
To determine the frequency and genomic location of recombination breakpoints among the four distinct HCoV species, we employed RDP5 which integrates multiple recombination algorithms in a single analysis. Our findings reveal that recombination in HCoV-229E alphacoronavirus is rare, with only two events detected in the spike–nucleocapsid region (Region 1 in Fig. 1a). This event comprised of two isolates from China in 2018 that were flagged as potential recombinants as they share mosaicism with two parental Chinese isolates detected in 2016 and 2017. In contrast, the other alphacoronavirus HCoV-NL63 displayed a greater frequency of recombination events (n = 16) with breakpoints starting at the beginning of ORF1a and spike genes (Fig. 1b and Supplementary Table 1). One notable recombination event was detected in 18 individual isolates across seven algorithms, with a starting breakpoint at nucleotide position 19,516 within the ORF1ab and an ending breakpoint at position 21,614 in the spike gene.

Recombination events were identified by RDP5 for a HCoV-229E, b HCoV-NL63, c HCoV-OC43, and d HCoV-HKU1. The resulting recombination regions flanked by a pair of breakpoints are indicated by coloured horizontal bars. Breakpoint regions are displayed according to their position relative to the annotated full genome of the virus. Inter-clade recombination event was observed for two distinct clades (HCoV-HKU1A and HCoV-HKU1B) of HCoV-HKU1 viruses. Maximum likelihood trees were reconstructed based on the regions before/after the breakpoints of Region 4 (blue dashed lines and orange asterisks) and Region 4 only (pink dashed lines). The bootstrap support values of ≥50 are shown above branches.
In the case of betacoronaviruses, HCoV-OC43 (n = 14) showed a higher frequency of recombination events compared to HCoV-HKU1 (n = 7) (Fig. 1c). Notably, recombination breakpoints in HCoV-OC43 primarily span genes encoding structural (HE, spike, E, and N) and accessory proteins (ORF2a, ORF4 and ORF8). Amongst all HCoV-OC43 isolates, thirty-seven were identified as homologous recombinants, with the beginning breakpoint located from position 24860 nt to 27628 nt (Region 10 in Fig. 1c) within the spike gene of HCoV-OC43. Recombination breakpoints for HCoV-HKU1 were detected within various genes, including ORF1ab, HE, spike, ORF4 and N regions. The resulting fragments flanked by these breakpoints vary in length, ranging from 360 to 15746 bp long.
Interestingly, inter-clade recombination was detected in HCoV-HKU1, with one recombination event occurring between the HCoV-HKU1A and HCoV-HKU1B clades (Region 4, denoted by pink horizontal bar in Fig. 1d). Phylogenetic analysis based on the left region of breakpoint (5’UTR–12067 nt) and the right region of breakpoint (21733 nt–3’UTR) clearly indicates that three isolates of HCoV-HKU1B (N5, N20 and N21 isolated from Hong Kong in 2004) clustered with clade HCoV-HKU1B (ML bootstrap=100%, left topology in Fig. 1d). In contrast, ML phylogeny based on Region 4 (12068–21732 nt) indicates three HCoV-HKU1B isolates (N16, N17 and N22 isolated from Hong Kong in 2004 and 2005) nested within the HCoV-HKU1A clade, with strong bootstrap of 100% (right topology in Fig. 1d). Our results indicate intratypic recombination between HCoV-HKU1A and HCoV-HKU1B.
Next, we reconstructed individual phylogenetic trees based on breakpoint-free genomic regions for each HCoV species. The HCoV-229E phylogeny, largely based on the ORF1a/1b regions, displayed ladder-like structure (Supplementary Fig. 2). In contrast, the phylogenies for HCoV-NL63 and HCoV-HKU1 clearly demonstrate multiple co-circulating clades (Supplementary Figs. 3–4). However, the scarcity of genomic sequences in last few decades and the removal of recombination breakpoints make the evolutionary references less informative, restricting our understanding of the underlying drivers contributing to these phylogenetic patterns. Therefore, we will focus on three main genes: spike, RdRp and nucleocapsid, where a higher number of sequences are available.
Recent lineage diversification of HCoV-229E and HCoV-OC43 viruses
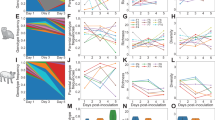
Temporal phylogenetic inference based on the spike gene indicates that HCoV-229E alphacoronaviruses have circulated in humans since the early 1960s (Fig. 2a). The global spike phylogeny of HCoV-229E can be arbitrarily classified into four genetic clades. The relative genetic diversity of HCoV-229E remained relatively constant for over 40 years (Fig. 2c) and mean effective reproduction number is below 1.0 during this early period (Fig. 2e). Since 2004, we observed frequent oscillations in genetic diversity, coinciding with a rapid lineage expansion of HCoV-229E from 2004 to 2010 (Fig. 2c), with relative genetic diversity reaching its peak around mid 2004 (Fig. 2c), while the corresponding Re rose from 1.0 to 1.5 (Fig. 2e). This period of expansion resulted in increased lineage diversification of HCoV-229E viruses, with the newer clade 3 viruses displacing clade 2, except for a unique case in the United States in 2016. Between 2011 to 2020 the HCoV-229E clade 3 viruses co-circulated with the derived clade 4 viruses, before apparently going extinct (Fig. 2a).

a, b Temporal phylogeographic spread of HCoV-229E and HCoV-OC43 during 1962–2022. Coloured branches represent ancestral location states. Amino acid mutations on the spike protein are mapped at major nodes of the trunk. The gray vertical bars indicate increased lineage diversification between 2004 and 2010. Posterior probability values of ≥0.95 are shown below branches. The inset indicates the duration of HCoV-229E persistence (in years) in different geographic locations. The dots indicate the estimated mean duration of persistence, and the horizontal bars represent the 95% highest posterior density (HPD) intervals. Red and blue stars denote new sequences from Singapore and Tanzania generated in this study. c, d Relative genetic diversity of HCoV-229E based on the spike gene, with the solid line representing the posterior mean values and the shaded area denoting the 95% HPD intervals e, f Estimates of the effective reproductive number of HCoV-229E over time. The posterior mean estimates with 95% intervals.
Notably, both HCoV-229E clade 3 and 4 viruses acquired significant non-synonymous amino acid mutations in the spike protein (Fig. 2a). Mapping these mutations onto a three-dimensional structure of the spike structure reveals that most mutations were concentrated in the RBD. Specifically, mutations Y305H, L310H, V325I, A323S, and Q349R are in the Domain B region (Fig. 3a, b). There are also mutations in Domain A (such as L90F, R89Q and V104A) and the S2 stalk region (R971I). These mutations suggest that the relative selection is greater on the RBD region, leading to genetic diversification and the emergence of new variants that facilitates the continuous circulation of HCoV-229E within humans. Our HCoV-229E temporal phylogeny indicates that six cases from Singapore (represented by red stars in Fig. 2a, Supplementary Fig. 2) grouped with clade 3 viruses were closely related to sequences from China and Hong Kong, while two Tanzanian cases (denoted by blue star in Fig. 2a) were related to sequences from multiple countries including USA, Japan and China. However, these isolates from Singapore and Tanzania were phylogenetically scattered, suggesting multiple independent introductions of HCoV-229E virus into these countries.

a Side view of the HCoV-229E trimeric spike (PDB ID: 6U7H) with major domains highlighted in colour. Domain A (38–267aa) is shown in cyan green; Domain B (293–435aa) in slate blue, with the receptor-binding domain (RBD; 297–434aa). The upper helix (UH; 629–652aa) is highlighted in purple, heptad repeat 1 (HR1; 795–871aa) in orange, and the central helix (CH; 873–918aa) in light green. b ribbon representation of the monomeric spike in HCoV-229E displaying significant amino acid mutations (marked by red spheres). Non-synonymous amino acid mutations identified as positively selected sites by MEME are highlighted in bolded and marked with asterisks. c Side view of the HCoV-OC43 trimeric spike (PDB ID: 6NZK) with coloured domains. Domain A (15–302aa); Domain B (335–607aa); UH: upper helix (913–930aa); HR1: heptad repeat 1 (1023–1086aa) and CH: central helix (1087–1137aa). d ribbon representation of the monomeric spike in HCoV-OC43 displaying significant amino acid mutations (marked by red spheres). Non-synonymous amino acid mutations identified as positively selected sites by MEME are highlighted in bolded and marked with asterisks. e Side view of the HCoV-NL63 trimeric spike (PDB ID: 7KIP) with coloured domains. Domain 0: (23–209aa); Domain A: (234–439aa); Domain B (481–616aa); RBD: receptor-binding domain (481–616aa). Domain 0 coloured in yellow; Domain A coloured in cyan green; Domain B in slate blue. f ribbon representative of the monomeric HCoV-NL63 spike displaying the amino acid mutations (marked by red spheres).
We further compared the relative genetic diversity of the RdRp and N genes and observed the population expansion of HCoV-229E between 2004–2010 was paired with a drop in genetic diversity that was most pronounced in the RdRp gene (Supplementary Fig. 5). However, after 2010, the genetic diversity of all three genes began to rise, reaching its peak in 2015. This increase was most prominent in the RdRp gene, coinciding with a marked increase in HCoV-229E cases in China and Hong Kong (Fig. 2). The recent decline in the genetic diversity of HCoV-229E during the 2020s is likely attributed to the stringent control measures during the COVID-19 pandemic that resulted in nearly zero circulation of other human respiratory viruses.
In comparison, the HCoV-OC43 betacoronavirus has been in circulation among humans for nearly 60 years, dating back to the 1960s (Fig. 2b). Prior to 2005, HCoV-OC43 viruses exhibited relatively consistent genetic diversity (Fig. 2d) and maintained a mean Re below 1.0 (Fig. 2f) for approximately 40 years, a similar pattern to HCoV-229E. From 2005 onwards there was a marked epidemic expansion of HCoV-OC43 viruses, accompanied by increased fluctuations in relative genetic diversity. Our isolates from Singapore (n = 8) were grouped into clades 3 and 4, while isolates from Tanzania (n = 3) fell into clades 2 and 4, capturing the broad genetic diversity of HCoV-OC43. Between 2005 and 2015, there was a significant rise in Re from less than 0.5 to 1.4. The patterns of relative genetic diversity of the RdRp and N genes are similar (Supplementary Fig. 6). Multiple HCoV-OC43 clades co-circulated since 2002, albeit clade 2 and 3 viruses have not been detected since 2019 and 2017, respectively, while clade 4 continues to circulate. The HCoV-OC43 virus has undergone significant lineage diversification and acquired a series of prominent non-synonymous amino acid mutations in the spike protein (Fig. 2). These non-synonymous mutations are predominantly found in both Domain A and B of the spike protein (Fig. 3c). Specifically, key mutations P22T, T25P, and K90L are in the N-terminal domain, which is responsible for binding to host sialic acids for viral entry. There is a notable cluster of spike mutations in Domain B, including V475K, T479A, F482L, N484D, and S486D, contributing to increased genetic divergence (Fig. 3d). Our analysis of the ratio of the number of non-synonymous and synonymous substitutions per site (dN/dS) indicates that the spike proteins of HCoV-229E and HCoV-OC43 (dN/dS = 0.393 and 0.356, respectively) were subject to reduced purifying selection than that of HCoV-NL63 (dN/dS = 0.164) and HCoV-HKU1 (dN/dS of HCoV-HKU1A = 0.230 and HCoV-HKU1B = 0.187), consistent with our global SLAC analysis (Table 2). The spike proteins of HCoV-229E and HCoV-OC43 also displayed greater dN/dS than other genes RdRp (HCoV-229E: 0.120; HCoV-OC43: 0.123) and N (HCoV-229E: 0.318; HCoV-OC43: 0.369).
The mean evolutionary rates of HCoV-229E and HCoV-OC43 based on the spike gene are estimated to be 7.34 × 10−4 and 8.79 × 10−4 substitutions/site/year, respectively (Table 3). These rates are also relatively faster than the estimated means for RdRp (229E: 2.54 × 10−4; OC43: 2.59 × 10−4 substitutions/site/year) and N (229E: 5.99 × 10−4; OC43: 5.15 × 10−4 substitutions/site/year; Supplementary Table 2). These inferred rates are broadly consistent with previous estimates20,21,23. The mean TMRCA dates of HCoV-229E and HCoV-OC43 were estimated as June 1961 and October 1964, respectively. From 1960 to the late 1980s, HCoV-229E viruses exhibited extended persistence (over 10 years) in Australia (Fig. 2a, Supplementary Table 3). However, the later HCoV-229E viruses showed shorter persistence in most countries, lasting around six months to 5 years (Fig. 2a). Similarly, the regional persistence of HCoV-OC43 virus exhibiting a similar pattern (Fig. 2b), typically less than five years in all observed geographic regions except China. Taken together, both HCoV-229E and HCoV-OC43 viruses are subject to more pronounced population bottlenecks, leading to the emergence of genetic variants and lineage turnover. In 2020, both viruses exhibited a decline in Re, likely attributed to the enforcement of stringent control measures during the COVID-19 pandemic.
Co-circulation and persistence of HCoV-NL63 and HCoV-HKU1 lineages
HCoV-NL63 viruses exhibited a slightly slower rate of evolution in the spike, at an estimated mean of 5.75 × 10−4 substitutions/site/year, compared to those observed for HCoV-229E and HCoV-OC43 (Table 3). This spike rate of NL63 is moderately faster than those of the RdRp and N genes (2.14 × 10−4 and 2.92 × 10−4 substitutions/site/year respectively; Supplementary Table 2), with the same magnitude as previously described24. In comparison, the HCoV-HKU1A spike (4.39 × 10−5 substitutions/site/year substitutions/site/year) evolves relatively slower than HCoV-HKU1B (1.46 × 10−4 substitutions/site/year). The mean TMRCA of HCoV-NL63 were estimated as June 1981, but HCoV-HKU1 was dated earlier in October 1954 and February 1951 for HCoV-HKU1A and HCoV-HKU1B clades, respectively. HCoV-NL63 was undetected in the United States until its first isolation and identification in the Netherlands in 2004, despite its presence as early as 198325,26. During the early evolution of HCoV-NL63, the virus segregated into two clades, referred to as clade 1 and 2 (Fig. 4a). Clade 1 viruses exhibited at least 24 non-synonymous amino acid mutations (Fig. 4a), however, these viruses stopped circulation in 2018. HCoV-NL63 clade 2 viruses demonstrated sequential non-synonymous amino acid mutations at the trunk of phylogeny, which diversified into smaller subclades, with clade 2.2 and 2.3 viruses still circulating in the population (Fig. 4a). We detected five cases from Singapore and three cases from Tanzania, all of which belong to multiple genetic lineages, indicating multiple importations of HCoV-NL63. Notably, HCoV-NL63 clade 2 viruses have accumulated several non-synonymous mutations, primarily concentrated in the more conserved S2 subunit of the spike (Fig. 3e). These mutations include R863H, S1086G, and V1095I. Five mutations (L25I, I49F, V84A, G109S and T110R) were identified within the distinctive 209-amino acid Domain 0 (Fig. 3f). This domain is unique to HCoV-NL63 and is a highly variable region, which plays a crucial role in immune escape27. Domain 0 binds to heparin sulfate, an attachment factor preceding spike-receptor engagement28. Comparatively, few mutations (e.g., H503R, I507L and A572E) targeted the antigenic RBD region of S1, suggesting that HCoV-NL63 may undergo weaker drift that resulted in the co-circulation of genetic lineages. This contrasts with HCoV-229E and HCoV-OC43 viruses where most non-synonymous mutations are located in Domains A and B of the spike.

a Temporal phylogeographic spread of HCoV-NL63 during 1983–2021. Coloured branches represent ancestral location states. Amino acid mutations on the spike protein are mapped at major nodes of the trunk. Posterior probability values of ≥0.95 are shown below branches. The inset indicates the duration of HCoV-NL63 persistence (in years) in different geographic locations. The dots indicate the estimated mean duration of persistence, and the horizontal bars represent the 95% highest posterior density (HPD) intervals. Red and blue stars denote new sequences from Singapore and Tanzania generated in this study. b Relative genetic diversity of HCoV-NL63 based on the nucleocapsid (shaded red), RdRp (shaded green) and spike genes (shaded orange), with the solid lines representing the posterior mean values and the shaded areas denoting the 95% HPD intervals c Estimates of the effective reproductive number of HCoV-NL63 over time. The posterior mean estimates with 95% intervals. d Correlation coefficient between pairwise genes (spike, nucleocapsid, and RdRp). e Estimates of time-series distances between the temporal population dynamics of one gene and another were inferred by the dynamic time warping (DTW) algorithm.
The relative genetic diversity of all three genes (RdRp, spike, and N) in HCoV-NL63 showed a gradual increase until the early 2000s (Fig. 4b). The genetic diversity of the N gene peaked around 2001, whereas the RdRp gene peaked around 2003, indicating the patterns of genetic diversity were not synchronized across the three genes. Additionally, a shift in geographical spread was observed from the United States to other locations, with multiple divergent clades emerging in Asia and Africa. The diversity of the N gene had declined approximately three-fold by 2005, with a less pronounced reduction in the RdRp and spike genes (Fig. 4b).
We further observed pronounced fluctuations in the effective reproductive number of HCoV-NL63, which was >1 in the 1980s then followed by a reduction to approximately 0.5 in the early 1990s (Fig. 4c). From 2006 to 2010, there was a notable rise in Re for HCoV-NL63 that coincided with a slight increase in genetic diversity in the spike gene but a decrease in genetic diversity of the N gene. Subsequently, the Re of HCoV-NL63 declined while genetic diversity remained stagnant until a second peak in Re occurred from 2014–2017. This final peak in Re corresponded with a drop in observed genetic diversity, and Re declined to below 1.0 up to the present day. The persistence of HCoV-NL63 viruses varied over time, ranging from 2.5 to 15 years, with longer persistence observed in the United States and China (Fig. 4a).
HCoV-HKU1 evolved into two genetically distinct lineages, designated HCoV-HKU1A and HCoV-HKU1B, shortly after its introduction into humans (Fig. 5a). The nucleotide similarities in the spike gene between HCoV-HKU1A and HCoV-HKU1B clades range from 83.9–86.0%, compared to within clade sequence similarities of 98.9–100% (HCoV-HKU1A) and 96.1–100% (HCoV-HKU1B). The HCoV-HKU1A lineage evolves at the slower rate (4.39 × 10−5 substitutions/site/year) compared to that of the HCoV-HKU1B lineage (Table 3). The TMRCA for HCoV-HKU1A and HCoV-HKU1B was estimated at 1954 and 1951, respectively. The HCoV-HKU1A lineage experienced an exponential increase in genetic diversity from the 1980s till the mid-2000s, followed by a decline in 2005, but peaked again in 2015 (Fig. 5b). This initial increase in diversity may be attributed to the widespread community transmission of HCoV-HKU1A viruses within Hong Kong, with a rise in Re to 2 in the early 2000s (Fig. 5c). A second increase in genetic diversity occurred in the early 2010s, which may be associated with increased HCoV-HKU1A infection in the United States and China.

a Temporal phylogeographic spread of HCoV-HKU1 showing two divergent clades (HCoV-HKU1A and HCoV-HKU1B). Coloured branches represent ancestral location states. Amino acid mutations on the spike protein are mapped at major nodes of the trunk. Posterior probability values of ≥0.95 are shown below branches. The inset indicates the duration of HCoV-HKU1 persistence (in years) in different geographic locations. The dots indicate the estimated mean duration of persistence, and the horizontal bars represent the 95% highest posterior density (HPD) intervals. Red and blue stars denote new sequences from Singapore and Tanzania generated in this study. b Relative genetic diversity of clade HCoV-HKU1A and HCoV-HKU1B viruses based on the nucleocapsid (shaded red), RdRp (shaded green) and spike genes (shaded orange), with the solid lines representing the posterior mean values and the shaded areas denoting the 95% HPD intervals c Estimates of the effective reproductive number of clade HCoV-HKU1A and HCoV-HKU1B viruses over time. The posterior mean estimates with 95% intervals.
In marked contrast, HCoV-HKU1B viruses have exhibited relatively constant genetic diversity over the last 60 year (Fig. 5b). Fewer amino acid mutations on the spike protein were detected that are primarily located in Domain A and B (Fig. 5a). Notably, the Re of HCoV-HKU1B viruses remained close to 1 for 40 years until 2000, when a sharp increase to nearly 4.0 occurred (Fig. 5c). Re showed a subsequent decline and was sustained at around 1 from 2010 to 2020.
Unlike other HCoVs, HCoV-HKU1 viruses showed markedly longer regional persistence geographically, variably from 8 to 59 years (Fig. 5a). Both HCoV-HKU1A and HCoV-HKU1B lineages do not display distinct seasonal patterns. The new sequences from Singapore and Tanzania are scattered throughout the phylogeny, indicative of multiple importations from other regions. Collectively, HCoV-HKU1 viruses are characterized by a slower evolutionary rates and antigenic drift, resulting in the co-circulation of multiple genetic lineages that persist for longer periods.
Correlation of time-series population dynamics among HCoV genes
To assess potential topological consistency between viral genes among different HCoV species, we reconstructed tanglegrams between paired phylogenies of RdRp, spike, and N. Our results indicate that the early HCoV-229E viruses showed relatively congruent phylogenies between spike and RdRp, as well spike and nucleocapsid, but later HCoV-229E viruses showed a mild level of incongruence between spike and RdRp, and spike and N (Fig. 6). For instance, a HCoV-229E isolate collected from Haiti, which belongs to clade 3 based on spike phylogeny, clustered with clade 4 viruses based on RdRp phylogeny, indicating the spike and RdRp may be derived from different sources. In comparison, HCoV-OC43, HCoV-NL63 and HCoV-HKU1 showed greater level of incongruence between spike and RdRp, compared to spike and N (Fig. 6).

For each HCoV species, maximum likelihood trees were inferred based on individual genes: RNA-dependent-RNA-polymerase (RdRp), spike (S) and nucleocapsid (N). Connecting lines line the same virus isolates across these genes. Coloured tips denote geographic locations.
To quantify the level of incongruence in genetic diversity among genes and species, we assessed the linear correlation of pairwise relative genetic diversity of the RdRp, spike and N for each HCoV. Both HCoV-229E and HCoV-NL63 viruses displayed a higher correlation coefficient (R2) between spike and RdRp genes, with respective R2 of 0.76 and 0.81 respectively, compared to other pairwise combinations (Fig. 4d). Conversely, HCoV-OC43 and HCoV-HKU1 showed a stronger correlation coefficient between the N and RdRp genes, with R2 of 0.75 and 0.98, respectively. Across all species, correlations between pairwise combinations of genes were generally below 0.5, indicating that the patterns in genetic diversity can vary among genes.
We used dynamic time warping (DTW) to further assess the time-series similarities between the temporal population variations of one gene and another. The total time-series distance (d) was calculated to assess all distances between two time points of pairwise genetic diversities throughout the study period, accounting for the temporal offset in epidemic peak values by allowing time scaling. The smaller the total time-series distance, the greater the similarity between two temporal patterns of genetic diversity. Specifically, smaller normalized distances among any three pairwise genes were observed for HCoV-229E (d: 0.05–1.57), suggesting the temporal epidemic patterns across these genes are similar (Fig. 4e). The HCoV-OC43 viruses also yielded smaller time-series distances (d: 1.02–1.58), indicative of greater similarities in population dynamics among genes temporally.
In contrast, HCoV-NL63 viruses displayed a greater time-series distance (d: 3.29–7.69) while HCoV-HKU1 viruses demonstrated the highest distances (d: 14.4–51.4) between pairwise gene combinations (Fig. 4e). This suggests that heterogeneity in population dynamics among genes for HCoV-NL63 and HCoV-HKU1, and this variation may be attributed to the frequencies and occurrence of recombination events (see above). Notably, the HCoV-HKU1 viruses also displayed higher levels of genetic diversity, with a mean ranging from 3.1–4.8, compared to the rest (mean genetic diversity of HCoV-229E: 1.6–2.0, HCoV-OC43: 2.5–2.9; HCoV-NL63: 2.9–3.0) (Fig. 5b), possibly due to the divergent evolutionary trajectories of the HCoV-HKU1A and HCoV-HKU1B lineages. These findings indicate that the inferred DTW distances between pairwise genes are reflective of the overall observed diversity among genes over time. As such, the DTW algorithm may serve as a robust virus transmission model for enabling real-time monitoring of epidemic dynamics at the population level.
Spatiotemporal transmission of human seasonal coronaviruses
We reconstructed the spatial dissemination of each of the four HCoV species following their introduction into humans. Our phylogeographic reconstruction based on the spike gene reveals that the source of HCoV-229E viruses was predominantly Australia from 1960–1980 with dissemination into the United States by the mid-1980s (Fig. 7a). Since 2000, we observed a global mixing of HCoV-229E in multiple regions including Hong Kong, China, and Italy (Fig. 7a), indicating widespread transmission of HCoV-229E viruses. Specifically, 28 strongly supported spatial diffusion pathways were observed between locations, such as from Hong Kong to China (BF > 13,000), China to Singapore (BF > 1025), from Australia into Hong Kong (BF = 181), and China to United States (BF = 78) (Fig. 7b, Supplementary Table 4). In comparison, HCoV-OC43 viruses were probably originated in the United States and France for a prolonged period (Fig. 7c). Since the 2000s, HCoV-OC43 viruses have spread into multiple geographic regions, including Canada, China, Japan, and Kenya. It was apparent that there is no single source of HCoV-OC43 diversity, as the proportion of ancestral areas at the phylogenetic trunk fluctuates from year to year. At least 41 strongly supported pathways were observed for HCoV-OC43 (Fig. 7d, Supplementary Table 5). Of these, eight disseminated out of China into several regions including Hong Kong, Kenya, Japan, the Netherlands, Singapore, Tanzania, United States and Vietnam.

a, c, e, g Trunk reward proportion plot through time for HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU1. b, d, f, h Supported migration flows from specific geographic locations to other locations for HCoV-229E, HCoV-OC43, HCoV-NL63 and HCoV-HKU1. The abbreviations used for the geographic location are as follows: Australia (AU), Belgium (BE), Brazil (BR), Canada (CA), China (CH), Côte d’Ivoire (CO), France (FR), Haiti (HA), Hong Kong (HK), Italy (IT), Japan (JP), Kenya (KY), Malaysia (MY), Mexico (MX), Netherlands (NL), Singapore (SG), South Korea (SK), Tanzania (TN), Thailand (TH), Uganda (UG), United Kingdom (UK), United States of America (USA), and Vietnam (VN).
During 1980–2005, HCoV-NL63 viruses appeared to have their possible source of transmission in the United States (Fig. 7e), despite the first reported human case originating in the Netherlands. Since 2005, China and the United States have become the main source for HCoV-OC43 viruses, although Japan had become more predominant in recent years (Fig. 7f). Notably, there are fewer supported diffusion pathways (13 pathways) observed for HCoV-NL63, with the majority originating from either China or the United States (Supplementary Table 6). Conversely, the source of HCoV-HKU1 transmission appeared to have originated from multiple regions (Fig. 7g), indicating that no single region was seeding HCoV-HKU1 epidemics. There were fewer transmission pathways (n = 16) observed for HCoV-HKU1 viruses (Fig. 7h, Supplementary Table 7). Strong migration support was observed from China to multiple locations (Australia, Hong Kong, Japan, Netherlands, and Thailand), from Hong Kong to Brazil and the United States, and from the United States to multiple locations (China, Hong Kong, Japan, and Singapore) (Supplementary Table 7).
To further assess the impact of sampling bias on spatiotemporal reconstruction, we conducted independent analyses using subsampled datasets for HCoV-229E, HCoV-OC43 and HCoV-NL63. The HCoV-HKU1 was excluded from the assessment of sampling bias due to limited number of sequences for the HKU1A and HKU1B clades. The ancestral location reconstructions, time persistence, trunk proportions and supported migration pathways were broadly corroborated between the subsampled and original datasets for HCoV-229E, HCoV-OC43 and HCoV-NL63 (Supplementary Figs. 7–11). For example, both datasets indicate that HCoV-229E consistently showed longer persistence in the early lineages in Australia, followed by intermixing of multiple regions in later HCoV-229E lineages (Supplementary Figs. 7–9). The patterns of trunk proportions over time in both datasets consistently indicate USA and China remained the primary sources for HCoV-OC43 and HCoV-NL63 (Supplementary Figs. 10 and 11).
Taken together, we show that both HCoV-229E and HCoV-OC43 viruses have experienced broader dissemination worldwide, whereas HCoV-NL63 and HCoV-HKU1 viruses exhibit more limited geographic spread. The enhanced geographic transmission observed for HCoV-229E and HCoV-OC43 may be attributed to faster evolutionary rates and greater selective pressure, leading to a higher number of non-synonymous substitutions concentrated in the spike RBD domain. These mutations likely lead to higher rates of antigenic drift that allows the viruses to evade host immune response and facilitating their spread.
Discussion
Four species of HCoVs have persistently circulated within humans for at least 60 years. The difficulty of culturing clinical HCoVs on commercial cell lines limits the understanding of the virus transmission characteristics and evolutionary behaviour. To the best of our knowledge, this study is among the first large-scale collaborative efforts involving clinicians and scientists in Singapore and Tanzania, screening and testing 3042 nasal swab samples from patients, that predates the COVID-19 pandemic. Unlike several published studies that rely solely on previously published genetic data, our study involves the direct collection and testing of clinical specimens. We infected differentiated human nasal epithelial cells with HCoV-NL63, HCoV-OC43 and HCoV-HKU1 viruses that were able to replicate at 33 °C six days post infection, thereby achieving a higher likelihood of yielding complete genomes. Our success allowed us to contribute an increase of 4.0–13.1% novel complete genomes for HCoV-229E (13.1%, 8/61), HCoV-NL63 (8.5%, 8/94), HCoV-OC43 (4.0%, 11/270) and HCoV-HKU1 (5.9%, 4/67). The integration of deep sequencing with robust evolutionary analyses has provided profound insights into the population dynamics and epidemic profiling of HCoVs.
We have revealed varying epidemic patterns and distinct phylodynamic profiles between HCoVs. We show that HCoV-229E and HCoV-OC43 viruses has a significant shift in epidemic expansion since 2004 with their effective reproductive number (Re) surpassing the threshold of 1. It is noteworthy that the increase in Re for HCoV-229E lasted for nearly five years followed by a Re decline in May 2009, coinciding with the timing of the pandemic emergence of influenza A/H1N1 virus in Mexico in early April 200929. By end April/early May 2009, Singapore had implemented control measures such as quarantine of incoming travellers from Mexico, isolation of suspected cases, contact tracing and aggressive testing for H1N1/2009 virus and full personal protective equipment in high-risk hospital units30. We found that HCoV-229E and HCoV-OC43 evolve at a faster evolutionary rate and are subject to reduced purifying selection pressure compared to HCoV-NL63 and HCoV-HKU1 viruses (see Table 4). We anticipate that the evolutionary trajectories of HCoV-229E and HCoV-OC43 will show more pronounced ladder-like spike phylogenies, where the older lineages go extinct, followed by regular replacement by newer lineages. This contrasts with the relatively lower dN/dS immune pressure and less pronounced drift observed in HCoV-NL63 and HCoV-HKU1 viruses which have extended co-circulation of multiple lineages.
HCoV-229E and HCoV-OC43 viruses have accumulated a series of significant amino acid substitutions at the trunk of their phylogenetic trees. These substitutions are mostly concentrated within the RBD of the spike protein, consistent with recent studies8,20,21. In HCoV-229E, the majority of amino acid substitutions occurred within the RBD Domain B of the S1 subunit, rather than Domain A, that potentially confers a selective advantage that enhances binding affinity for the APN receptor8. Previous studies have shown that serum samples from older HCoV-229E strains (pre-1990) are incapable of neutralizing newer HCoV-229E strains31, indicative of antigenic disparities. In comparison, the acquisition of non-synonymous mutations in HCoV-OC43 largely occurred in both Domain A and B regions. A recent study has indicated that while there are neutralising antibodies that target the sialoglycan-receptor binding site in Domain A, more neutralising antibodies target Domain B22, highlighting the potential importance of Domain B. Further experiments are required to examine the role of Domain B in virus entry. Consequently, these mutations in HCoV-OC43 viruses may lead to antigenic drift to escape from host immune recognition. Taken together, the recent emerging strains of HCoV-229E and HCoV-OC43 possess the ability to displace older circulating strains, echoing the evolutionary trend observed in human influenza A/H1N1 and A/H3N2 viruses32,33,34. Analogous to the spike protein, the influenza hemagglutinin protein experiences strong immune pressure to evade host immune response leading to continuous antigenic drift and frequent lineage replacement. We further observe that recombination events were more frequent in HCoV-NL63 and HCoV-OC43, with breakpoint regions occurring throughout HCoV-NL63 genomes, while breakpoints were primarily located in HCoV-OC43 structural genes. In contrast, HCoV-229E viruses do not undergo frequent recombination, consistent with previous observation35.
While HCoV-NL63 and HCoV-HKU1 viruses display similar epidemic behaviour and undergo weaker selection pressure, they differ in their phylodynamic patterns. HCoV-NL63 has a moderate evolutionary rate and exhibits mild temporal fluctuations in relative genetic diversity, in contrast to lower evolutionary rate and relatively constant genetic diversity observed in HCoV-HKU1. Temporal phylogenetics shows that HCoV-NL63 has multiple genetic clades that have co-circulated for several decades. We also observed that, unlike HCoV-229E and HCoV-OC43 viruses, HCoV-NL63 viruses exhibit limited mutations in the RBD region which likely contributes to slower antigenic evolution and lineage turnover. An experimental study using hNECs has shown that HCoV-NL63 viruses replicate less efficiently compared to HCoV-229E viruses36, consistent with our evolutionary analyses, and possibly explaining why HCoV-229E is more dominant and widespread than HCoV-NL63.
HCoV-HKU1 viruses have the lowest evolutionary rate amongst the four human coronaviruses, however the two distinct lineages (HCoV-HKU1A and HCoV-HKU1B) demonstrate differing phylodynamics, with HCoV-HKU1A having a steady rise in relative genetic diversity while HCoV-HKU1B showed constant genetic diversity. Both clades had a substantial number of amino acid substitutions in the spike, but these mostly occurred outside the RBD. Furthermore, HCoV-229E spike exhibits significantly lower number of positively selected sites compared to HCoV-229E, HCoV-NL63 and HCoV-OC43, suggesting that genetic drift is not a prominent factor driving the evolution of HCoV-HKU1. This may have contributed to the co-existence of two divergent HCoV-HKU1A and HCoV-HKU1B clades, without apparent lineage turnover. The evolutionary pattern of HCoV-HKU1 bears resemblance to that of the human parainfluenza 4 viruses, which similarly consists of two genetic clades, HPIV4A and HPIV4B, that have only limited circulation worldwide37. Additionally, HCoV-HKU1 exhibits moderate recombination events although inter-clade recombination occurs between HCoV-HKU1A and HCoV-HKU1B viruses. Recombination may play a significant role in facilitating wider genetic divergence at the population level, thereby contributing to the longer-term persistence and successful survival of HCoV-HKU1.
Our Bayesian phylogeographic inference suggests that ancestral HCoV-229E and HCoV-OC43 viruses primarily occupied a single geographic region, with HCoV-229E originating predominantly from Australia and HCoV-OC43 from the United States. However, phylogeographic findings should be interpreted with caution, particularly with the limited availability of HCoV sequences prior to the 2000s, and the uneven geographic and temporal distribution of samples from Africa, Southeast Asia, and South America. Despite these limitations, we observed that later strains of HCoV-229E and HCoV-OC43 viruses show a notable shift indicating multiple geographic regions as potential sources. These viruses may have facilitated global dissemination and intermixing across multiple geographic locations through air travel, such as China, Hong Kong, Japan, Europe, and Southeast Asia. Conversely, HCoV-NL63 and HCoV-HKU1 viruses show less frequent international transmission, with sustained local spread being more common.
We utilized linear regression and dynamic time warping (DTW) methods to understand the relationship between the genetic diversity of one gene and another. Our findings suggest that one gene alone cannot predict the diversity of other genes, as recombination, and possibly differential natural selection pressures, complicates the complexity of HCoV evolution. The DTW algorithm and similar platforms have been developed to compare the time-series relationship among different parameters for SARS-CoV-238,39,40, including the incidence of new cases, hospital admission number, climatic factors, and human mobility. This tool serves as a valuable application in forecasting future epidemic growth and transmission of different infectious diseases.
The COVID-19 pandemic has provided an exceptional opportunity for studying molecular epidemiology, evolution, and the vaccine effectiveness of SARS-CoV-2 virus. Through global surveillance and sequencing efforts, we have gained insights into the underlying mechanisms driving the emergence and rapid displacement of novel variants that have led to successive waves of global outbreaks41,42,43,44. This is mostly driven by the accumulation of key mutations in the spike protein of SARS-CoV-2, with experimental evidence that these mutations are associated with increased infectivity and transmission45,46,47. We speculate that HCoV-229E and HCoV-OC43 will undergo continuous antigenic evolution, driven by the spike mutations, thereby driving clade diversification, and shaping their evolutionary trajectories over time. This study also reveals that differential spike mutations among these four HCoV species, highlighting their specific binding preferences to host receptors. Future experiments should be designed to comprehend the biological impact of significant mutations in HCoVs, particularly relating to potential changes in receptor binding affinity, growth kinetics, duration of shredding and pathogenesis both in vitro and in vivo, particularly using hNECs. This platform will offer a more comprehensive understanding of the biological characteristics, including replicative fitness and pathogenesis, by mimicking HCoV infections in the natural human nasal epithelial environment. With the increasing demand for metagenomic next-generation sequencing, we anticipate that more genomic data of HCoVs will become available and coupled with machine learning algorithms, the future trajectories of these viruses will be more accurately predicted. While no vaccines are currently available for HCoVs, preventive strategies such as the incorporation of virus strains in the COVID-19 vaccines should be considered to potentially reduce the severity of pneumonia, intensive care unit admission as well as rates of hospitalisation. Similar to influenza vaccines, which incorporate three types of seasonal influenza (A/H1N1, A/H3N2 and B/Victoria), this approach could be explored for HCoV vaccination to reduce the spread of coronaviruses and risk of severe illness.
Methods
Ethics statement
This study was approved by the ethics committees at the National University of Singapore Institutional Review Board (NUS-IRB: B-14-209E and H-18-071E), Singapore. Nasal biopsies were collected from different patients who underwent septal plastic surgery at the National University Hospital (NUH), Singapore (NUS-IRB: L13-509). Written informed consent was obtained from all adult participants and all clinical samples were deidentified.
Virus samples, culture, and sequencing
A total of 3042 nasopharyngeal swab samples were collected from adult patients in Singapore48 during 2007–2013 and Tanzania during 2016–2018. Identification of positive samples for a plethora of common human respiratory viruses (including HCoVs) followed our previous study in Singapore49. All HCoV-229E virus samples were cultured in Caco2 cells following our published methods50. The remaining HCoV-NL63, HCoV-OC43, and HCoV-HKU1 samples were cultured using fully differentiated human nasal epithelial cells (hNECs) through an air-liquid interface (ALI). The biopsy samples were differentiated into hNECs as previously described in ref.51. Fifty microliters of individual virus samples were inoculated onto hNECs and incubated for 10 min at 33 °C. The inoculum was removed and the infected hNECs were incubated at 33 °C for 6 days. At 6 dpi (days post-inoculation), 100 µl of phosphate buffered saline (PBS) were added to each transwell and incubated with the infected cells for 10 min at 33 °C. Apical washes were collected and stored at −80 °C. Viral RNA was then extracted from the supernatant (only for HCoV-229E) or apical washes using the Direct-zol RNA MiniPrep kit (Zymo Research, USA). The CT values of cultured samples were measured by RT-quantitative PCR following published protocols52. Full genome sequencing of HCoVs were performed using next-generation sequencing. Briefly, extracted RNA was used for direct RNA library preparations with the TruSeq RNA Library Prep Kit or RNA Prep with Enrichment (L) Tagmentation (Illumina International, USA) according to the manufacturer’s instruction. The quality of the libraries was verified using the Agilent Bioanalyzer (Agilent Technologies, USA). Libraries were pooled and run an Illumina Miseq Platform using a read length of 2 × 250 bp. The raw NGS reads were quality-checked and trimmed using Trimmomatic v0.3953 to remove adaptors and low-quality reads. Consensus genomic sequences were assembled using the BWA-MEM algorithm54 in UGENE v.3455. We obtained 31 complete genomes for HCoV-229E (n = 8), HCoV-NL63 (n = 8), HCoV-OC43 (n = 11) and HCoV-HKU1 (n = 4).
Recombination analysis
We downloaded all available global sequences from NCBI GenBank for all four HCoV species from 1960 to 2023. For each HCoV species, recombination analysis was performed based on complete genomes (HCoV-229E: n = 61; HCoV-NL63: n = 94; HCoV-OC43: n = 270; and HCoV-HKU1: n = 106) using RDP5 software56. The occurrence of putative recombination events was detected using multiple algorithms: RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan, 3Seq and LARD. The criteria for valid recombination events were determined as follows: at least two virus isolates, at least five different algorithms and P-value < 0.05. The detection of recombination regions and corresponding breakpoint locations at the beginning and the end were indicated in Fig. 1.
Phylodynamics and phylogeographic analysis
Individual datasets were curated for HCoV-229E based on RdRp (n = 134), spike (n = 162), and nucleocapsid sequences (n = 187) and breakpoint-free genomic regions (n = 61). The HCoV-NL63 datasets are RdRp (n = 151), spike (n = 141), nucleocapsid sequences (n = 209) and breakpoint-free genomic regions (n = 94). The HCoV-OC43 datasets have RdRp (n = 306) and spike (n = 589), nucleocapsid (n = 284), and breakpoint-free genomic regions (n = 270), The HCoV-HKU1 datasets have RdRp (n = 118) and spike (n = 106), nucleocapsid (n = 150) and breakpoint-free genomic regions (n = 68). The temporal signal was examined using TempEst57 and the root-to-tip analysis of HCoV-229E and HCoV-NL63 spike sequences showed stronger temporal signal (R2 = 0.97 and 0.63, respectively) compared to HCoV-OC43, HCoV-HKU1A and HCoV-HKU1B (R2 = 0.22–0.34). However, a previous study has indicated significant discrepancies in R2 depending on the number of sequences included and the year of collection21. For each individual gene (spike, RdRp and N), population dynamics were estimated independently for HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1, using an uncorrelated lognormal (UCLN) relaxed clock, General Time Reversible (GTR) substitution model with a gamma (G) distribution of among-site rate variation (I) and a Gaussian Markov Random Field (GMRF) Bayesian skyride model using BEAST v1.10.458. The uncorrelated lognormal relaxed clock and GTR + G + I has been recommended for the estimation of substitution rates1,21,59,60. The UCLN relaxed clock and the GTR + G + I substitution model have been widely recommended for estimating substitution rates in rapidly evolving viruses1,21,59,60, as they account for the natural variability in evolutionary rates across viral lineages. In the spike analysis of HCoV-HKU1, our preliminary analysis using the UCLN model did not achieve convergence, since the spike phylogeny of HCoV-HKU1 virus is composed of two genetically divergent clades (clade 1A and clade 1B). We further applied a fixed local clock model61 to estimate the rates of evolution and population dynamics simultaneously for two independent genetic lineages that are strictly monophyletic. The use of a fixed local clock model allows the lineage- or host-specific clock model to estimate rates of evolution in different magnitude, which cannot be achieved with the UCLN model.
The spatial-temporal patterns of each HCoV species were inferred using a discrete-state continuous-time Markov model with the Bayesian Stochastic Search Variable Selection procedures, where geographic locations were coded as discrete states. For each dataset, four independent runs of Bayesian Markov Chain Monte Carol (MCMC) chains were performed, each with 100 million generations and sampling every 10,000 generations (virus isolates are listed in Supplementary Tables 8–11). The convergence was checked with effective sample size (ESS) values of at least 200 and appropriate burn-in was applied. The resulting log and tree files were combined using LogCombiner v1.10.4. The rates of nucleotide substitution, the time to most recent common ancestor (TMRCA), and the past population dynamics of each species were estimated simultaneously. The relative diversity over time represented as the effective population size was inferred within Tracer v.1.7.2 using the GMRF smoothing prior. The resulting output files were plotted in ggplot262 in R. We assessed the relative genetic diversity of the larger and smaller datasets for HCoV-229E, HCoV-NL63 and HCoV-OC43, the relative genetic diversities were not severely impacted by the sampling density (Supplementary Fig. 12). Maximum clade credibility (MCC) trees were generated using TreeAnnotator. Phylogenies based on non-recombinant regions for HCoV-229E, HCoV-NL63 and HCoV-HKU1 are shown in Supplementary Figs. 2–4, however HCoV-OC43 where analyses based on non-recombinant regions indicated a much slower evolutionary rate. This slower rate implies that long divergence times and inadequate sampling may produce a false recombination signal. The removal of recombination breakpoint regions can make the evolutionary inferences less informative and robust.
Discrete phylogeographic reconstructions and spatial diffusion were inferred based on spike gene for each species. The Bayes Factor of diffusion pathways showing the spatiotemporal spread of human coronaviruses was calculated using SpreaD363. Significant diffusion pathways were inferred based on Bayes Factors (BF): BF ≥ 1,000 was deemed as decisive support, 100 ≤ BF < 1000 as very strong support, 10 ≤ BF < 100 as strong support and 3 ≤ BF < 10 as supported64. The time persistence and trunk proportion for each geographic location were calculated using PACT v0.0.565. Sankey plot was generated to indicate diffusion pathways from one geographic location to another by ggsankey package in R. We also assessed the impact of sampling bias on the spatiotemporal diffusion patterns by subsampling three datasets for all species except HCoV-HKU1 due to insufficient data, using proportional sampling from an inferred maximum likelihood tree by smot66. The resulting phylogeographic reconstructions, time persistence and trunk proportions were compared.
Selection analysis
We compared the selection pressures among the four species of HCoVs based on our generated and global sequences of three individual genes (RdRp, spike and N) using HyPhy v.2.5.5967. The ratio of non-synonymous to synonymous substitutions (dN/dS) per site were estimated using Mixed Effects Model of Evolution (MEME)68 and Single Likelihood Ancestor Counting (SLAC)69 for detecting episodic and pervasive selection, respectively. Positively selected sites significant value at (P < 0.05) were identified.
Estimates of reproductive number
The effective reproductive number (denoted as Re) of each HCoV species was inferred based on the spike gene using BEAST 2 v.2.7.370. The estimates of Re is the average number of secondary infections caused by an infected individual in a population, which is a useful indicator for assessing the epidemic growth or the effectiveness of interventions. The Re was obtained by dividing transmission rate (λ) by the becoming noninfectious rate (δ). This parameterization is analogous to dividing the births of new infectious individuals by the termination or “death” of their infectious status. An Re > 1 at any point of time would indicate an expanding outbreak while an Re < 1 would indicate a receding outbreak. A birth-death skyline serial (BDSKY) model71 as implemented in BEAST 2 was applied to estimate Re of each HCoV species. For each species, an optimized relaxed clock model and a GTR nucleotide substitution model were used. The reproductive number was set to ten dimensions across the study period. A chain length of 100 million generations was performed using BEAST 2, with sampling at every 10,000 generations. Convergence of the runs were checked in Tracer v.1.7.2 and appropriate burn-in values were removed. The R package bdskytools was used to visualize the BDSKY results and plot varying Re over time.
Correlation of population dynamics among genes
Linear correlations of between the diversity of each gene per HCoV lineage was assessed with a simple linear model \(y={\beta }_{0}+{\beta }_{1}x\) using the lm function, where y represents the diversity of one gene, and \(x\) represents the the diversity of a second gene within the same dataset. For each HCoV species, the same isolates and same number of spike sequences were used as stated above. We also applied a dynamic time warping (DTW) algorithm to calculate the minimal distances between the genetic diversity of one gene and another as a time series per year by using the R package ‘dtw’72. For each HCoV species, normalized distances were calculated to compare the similarities between temporal population dynamics among pairwise genes.
Amino acid reconstruction and structural modelling
Amino acid mutations were reconstructed on the three-dimensional (3D) structure of the spike protein using the treesub program73 and mapped onto the phylogenies. PyMOL v2.3.574 was used to visualize and map the tree substitution mutations. The spike protein structures for HCoV-229E (6U7H)75, HCoV-NL63 (7KIP)76, and HCoV-OC43 (6NZK)77 were downloaded from Protein Data Bank (PDB).
Tanglegram reconstruction
To investigate the potential co-evolution among genes of human coronaviruses, maximum likelihood phylogenies of each individual gene (RdRp, spike and N) were constructed by RAxML-NG v.1.1.078. For each species, tanglegrams were visualised based on reconstructed maximum-likelihood gene trees and visualized using in-house Python scripts using libraries Matplotlib and baltic.
Data availability
New sequences have now been deposited to NCBI GenBank, with new accession numbers indicated in the manuscript.
References
Corman, V. M. et al. Evidence for an Ancestral Association of Human Coronavirus 229E with Bats. J. Virol. 89, 11858–11870 (2015).
Tao, Y. et al. Surveillance of Bat Coronaviruses in Kenya Identifies Relatives of Human Coronaviruses NL63 and 229E and Their Recombination History. J. Virol. 91, e01953–16 (2017).
Lau, S. K. P. et al. Discovery of a novel coronavirus, China Rattus coronavirus HKU24, from Norway rats supports the murine origin of Betacoronavirus 1 and has implications for the ancestor of Betacoronavirus lineage A. J. Virol. 89, 3076–3092 (2015).
Kanwar, A., Selvaraju, S. & Esper, F. Human Coronavirus-HKU1 Infection Among Adults in Cleveland, Ohio. Open Forum Infect. Dis. 4, ofx052 (2017).
Bastien, N. et al. Human Coronavirus NL63 Infection in Canada. J. Infect. Dis. 191, 503 (2005).
Shah, M. M. et al. Seasonality of common human coronaviruses, United States, 2014–2021. Emerg. Infect. Dis. 28, 1970–1976 (2022).
Edridge, A. W. D. et al. Seasonal coronavirus protective immunity is short-lasting. Nat. Med. 26, 1691–1693 (2020).
Forni, D. et al. Adaptation of the endemic coronaviruses HCoV-OC43 and HCoV-229E to the human host. Virus Evol. 7, veab061 (2021).
Cui, J., Li, F. & Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol 17, 181–192 (2019).
Zeng, Q., Lan gereis, M. A., van Vliet, A. L. W., Huizinga, E. G. & de Groot, R. J. Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc. Natl. Acad. Sci. USA 105, 9065–9069 (2008).
Yeager, C. L. et al. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357, 420–422 (1992).
Hofmann, H. et al. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 102, 7988–7993 (2005).
Hulswit, R. J. G. et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc. Natl. Acad. Sci. USA 116, 2681–2690 (2019).
Saunders, N. et al. TMPRSS2 is a functional receptor for human coronavirus HKU1. Nature 624, 207–214 (2023).
Sola, I., Almazán, F., Zúñiga, S. & Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu Rev. Virol. 2, 265–288 (2015).
Müller, N. F., Kistler, K. E., & Bedford, T. A.Bayesian approach to infer recombination patterns in coronaviruses. Nat. Commun. 13, 4186 (2022).
Anthony, S. J. et al. Further Evidence for Bats as the Evolutionary Source of Middle East Respiratory Syndrome Coronavirus. mBio 8, e00373–17 (2017).
Pyrc, K. et al. Culturing the Unculturable: Human Coronavirus HKU1 Infects, Replicates, and Produces Progeny Virions in Human Ciliated Airway Epithelial Cell Cultures. J. Virol. 84, 11255–11263 (2010).
Farsani, S. M. J. et al. The first complete genome sequences of clinical isolates of human coronavirus 229E. Virus Genes 45, 433–439 (2012).
Kistler, K. E. & Bedford, T. Evidence for adaptive evolution in the receptor-binding domain of seasonal coronaviruses OC43 and 229e. eLife 10, e64509 (2021).
Jo, W. K., Drosten, C. & Drexler, J. F. The evolutionary dynamics of endemic human coronaviruses. Virus Evol. 7, veab020 (2021).
Wang, C. et al. Antigenic structure of the human coronavirus OC43 spike reveals exposed and occluded neutralizing epitopes. Nat. Commun. 13, 2921 (2022).
Lau, S. K. P. et al. Molecular Epidemiology of Human Coronavirus OC43 Reveals Evolution of Different Genotypes over Time and Recent Emergence of a Novel Genotype due to Natural Recombination∇. J. Virol. 85, 11325–11337 (2011).
Pyrc, K. et al. Mosaic Structure of Human Coronavirus NL63, One Thousand Years of Evolution. J. Mol. Biol. 364, 964–973 (2006).
Fouchier, R. A. M. et al. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. USA 101, 6212–6216 (2004).
van der Hoek, L. et al. Identification of a new human coronavirus. Nat. Med. 10, 368–373 (2004).
Abdul-Rasool, S. & Fielding, B. C. Understanding Human Coronavirus HCoV-NL63. Open Virol. J. 4, 76–84 (2010).
Milewska, A. et al. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol. 88, 13221–13230 (2014).
Smith, G. J. D. et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459, 1122–1125 (2009).
Tay, J., Ng, Y. F., Cutter, J. L. & James, L. Influenza A (H1N1-2009) pandemic in Singapore-public health control measures implemented and lessons learnt. Ann. Acad. Med. Singap. 39, 313–312 (2010).
Eguia, R. T. et al. A human coronavirus evolves antigenically to escape antibody immunity. PLOS Pathog. 17, e1009453 (2021).
Bedford, T. et al. Global circulation patterns of seasonal influenza viruses vary with antigenic drift. Nature 523, 217–220 (2015).
Dhanasekaran, V. et al. Human seasonal influenza under COVID-19 and the potential consequences of influenza lineage elimination. Nat. Commun. 13, 1721 (2022).
Su, Y. C. F. et al. Phylodynamics of H1N1/2009 influenza reveals the transition from host adaptation to immune-driven selection. Nat. Commun. 6, 7952 (2015).
Pollett, S. et al. A comparative recombination analysis of human coronaviruses and implications for the SARS-CoV-2 pandemic. Sci. Rep. 11, 17365 (2021).
Otter, C. J. et al. Infection of primary nasal epithelial cells differentiates among lethal and seasonal human coronaviruses. Proc. Natl. Acad. Sci. USA 120, e2218083120 (2023).
Linster, M. et al. Clinical and Molecular Epidemiology of Human Parainfluenza Viruses 1-4 in Children from Viet Nam. Sci. Rep. 8, 6833 (2018).
Morel, J.-D., Morel, J.-M. & Alvarez, L. Time warping between main epidemic time series in epidemiological surveillance. PLOS Comput. Biol. 19, e1011757 (2023).
Lin, G. et al. Investigating the effects of absolute humidity and movement on COVID-19 seasonality in the United States. Sci. Rep. 12, 16729 (2022).
Ward, T., Johnsen, A., Ng, S. & Chollet, F. Forecasting SARS-CoV-2 transmission and clinical risk at small spatial scales by the application of machine learning architectures to syndromic surveillance data. Nat. Mach. Intell. 4, 814–827 (2022).
McCrone, J. T. et al. Context-specific emergence and growth of the SARS-CoV-2 Delta variant. Nature 610, 154–160 (2022).
Hodcroft, E. B. et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 595, 707–712 (2021).
Paton, R. S., Overton, C. E. & Ward, T. The rapid replacement of the SARS-CoV-2 Delta variant by Omicron (B.1.1.529) in England. Sci. Transl. Med. 14, eabo5395 (2022).
Markov, P. V. et al. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 21, 361–379 (2023).
Ulrich, L. et al. Enhanced fitness of SARS-CoV-2 variant of concern Alpha but not Beta. Nature 602, 307–313 (2022).
Saito, A. et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 602, 300–306 (2022).
Shi, G. et al. Omicron Spike confers enhanced infectivity and interferon resistance to SARS-CoV-2 in human nasal tissue. Nat. Commun. 15, 889 (2024).
Low, J. G. H. et al. Early Dengue infection and outcome study (EDEN) - study design and preliminary findings. Ann. Acad. Med Singap. 35, 783–789 (2006).
Chen, Y. et al. Etiology of febrile respiratory infections in the general adult population in Singapore, 2007–2013. Heliyon 7, e06329 (2021).
Mah, M. G. et al. Spike-independent infection of human coronavirus 229E in bat cells. Microbiol. Spectr. 11, e0348322 (2023).
Gamage, A. M. et al. Human nasal epithelial cells sustain persistent sars-cov-2 infection in vitro, despite eliciting a prolonged antiviral response. mBio 22, e0343621 (2022).
Hoek, R. A. S. et al. Incidence of viral respiratory pathogens causing exacerbations in adult cystic fibrosis patients. Scand. J. Infect. Dis. 45, 65–69 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Okonechnikov, K., Golosova, O., Fursov, M. & team, U. G. E. N. E. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167 (2012).
Martin, D. P. et al. RDP5: a computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 7, veaa087 (2021).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolut. Biol. 7, 214 (2007).
Shao, N. et al. Molecular evolution of human coronavirus-NL63, -229E, -HKU1 and -OC43 in hospitalized children in China. Front. Microbiol. 13, 1023847 (2022).
Lau, S. K. P. et al. Molecular Evolution of Human Coronavirus 229E in Hong Kong and a Fatal COVID-19 Case Involving Coinfection with a Novel Human Coronavirus 229E Genogroup. mSphere 6, e00819-20 (2021).
Worobey, M., Han, G.-Z. & Rambaut, A. A synchronized global sweep of the internal genes of modern avian influenza virus. Nature 508, 254–257 (2014).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, New York, 2016).
Bielejec, F. et al. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 33, 2167–2169 (2016).
Virk, R. K. et al. Divergent evolutionary trajectories of influenza B viruses underlie their contemporaneous epidemic activity. PNAS 117, 619–628 (2020).
Bedford, T. et al. Strength and tempo of selection revealed in viral gene genealogies. BMC Evol. Biol. 11, 220 (2011).
Arendsee, Z. W. et al. smot: A python package and CLI tool for contextual phylogenetic subsampling. J. Open Source Softw. 7, 4193 (2022).
Kosakovsky Pond, S. L. et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 37, 295–299 (2019).
Murrell, B. et al. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLOS Genet. 8, e1002764 (2012).
Kosakovsky Pond, S. L. & Frost, S. D. W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 22, 1208–1222 (2005).
Bouckaert, R. et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLOS Comput.Biol. 15, e1006650 (2019).
Stadler, T., Kühnert, D., Bonhoeffer, S. & Drummond, A. J. Birth–death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc. Natl Acad. Sci. 110, 228–233 (2013).
Giorgino, T. Computing and Visualizing Dynamic Time Warping Alignments in R: The dtw Package. J. Stat. Softw. 31, 1–24 (2009).
Tamuri, A. U. Treesub: annotating ancestral substitution on a tree. https://github.com/tamuri/treesub (2013).
Schrödinger, L. L. C. The PyMOL Molecular Graphics System, Version 1.8. (2015).
Li, Z. et al. The human coronavirus HCoV-229E S-protein structure and receptor binding. Elife 8, e51230 (2019).
Zhang, K. et al. A 3.4-Å cryo-electron microscopy structure of the human coronavirus spike trimer computationally derived from vitrified NL63 virus particles. QRB Discov. 1, e11 (2020).
Tortorici, M. A. et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 26, 481–489 (2019).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019).
Acknowledgements
The study was supported by the Duke-NUS Signature Research Programme funded by the Ministry of Health, Singapore. We thank Duke-NUS Medical School Genome Biology Facility for assistance with sequencing, and all submitters for access to their sequences in GenBank and GISAID databases.
Author information
Authors and Affiliations
Contributions
G.J.D.S. and Y.C.F.S. conceived and designed the study; M.G.M. and Z.Y. conducted experiments; V.P.M., J.A.C., M.P.R., E.E.O. and J.G.H.L. provided samples, M.G.M., M.A.Z., R.Z. and Y.C.F.S. performed phylodynamic analysis; M.A.Z. and R.Z. wrote in-house scripts; M.A.Z. and Y.C.F.S. designed the Figures; D.Y.W. provided primary human nasal epithelial cells; M.G.M, M.A.Z., G.J.D.S and Y.C.F.S. wrote the original draft. All authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mah, M.G., Zeller, M.A., Zhang, R. et al. Discordant phylodynamic and spatiotemporal transmission patterns driving the long-term persistence and evolution of human coronaviruses. npj Viruses 2, 49 (2024). https://doi.org/10.1038/s44298-024-00058-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44298-024-00058-w
