
Autophagy, Unfolded Protein Response, and Neuropilin-1 Cross-Talk in SARS-CoV-2 Infection: What Can Be Learned from Other Coronaviruses

 , , , , ,
, , , , ,  , , ,
, , ,
Abstract
:1. Introduction
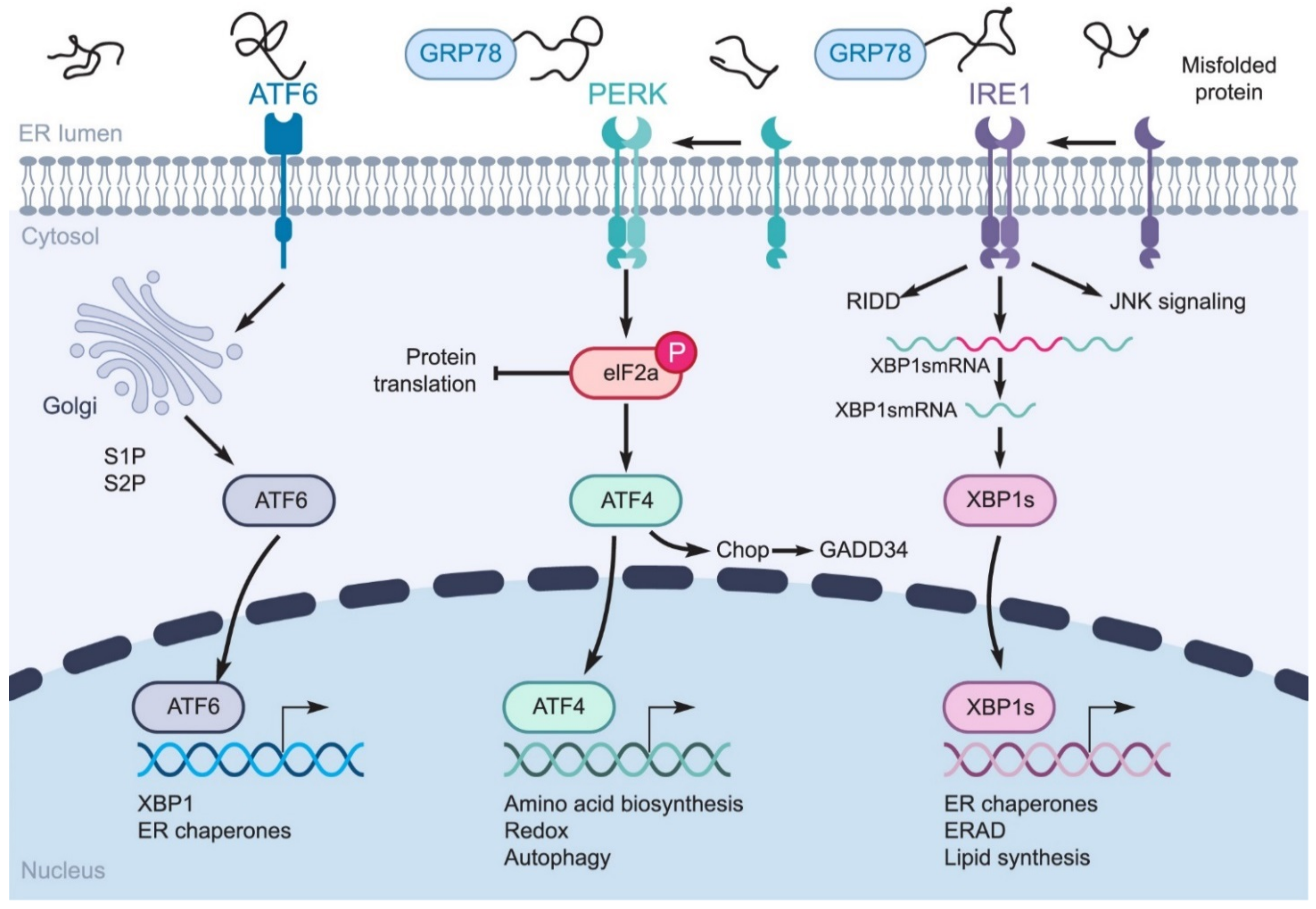
2. Coronavirus-Induced ER Stress Response Activates the UPR
2.1. GRP78 Facilitates Surface Coronavirus Attachment
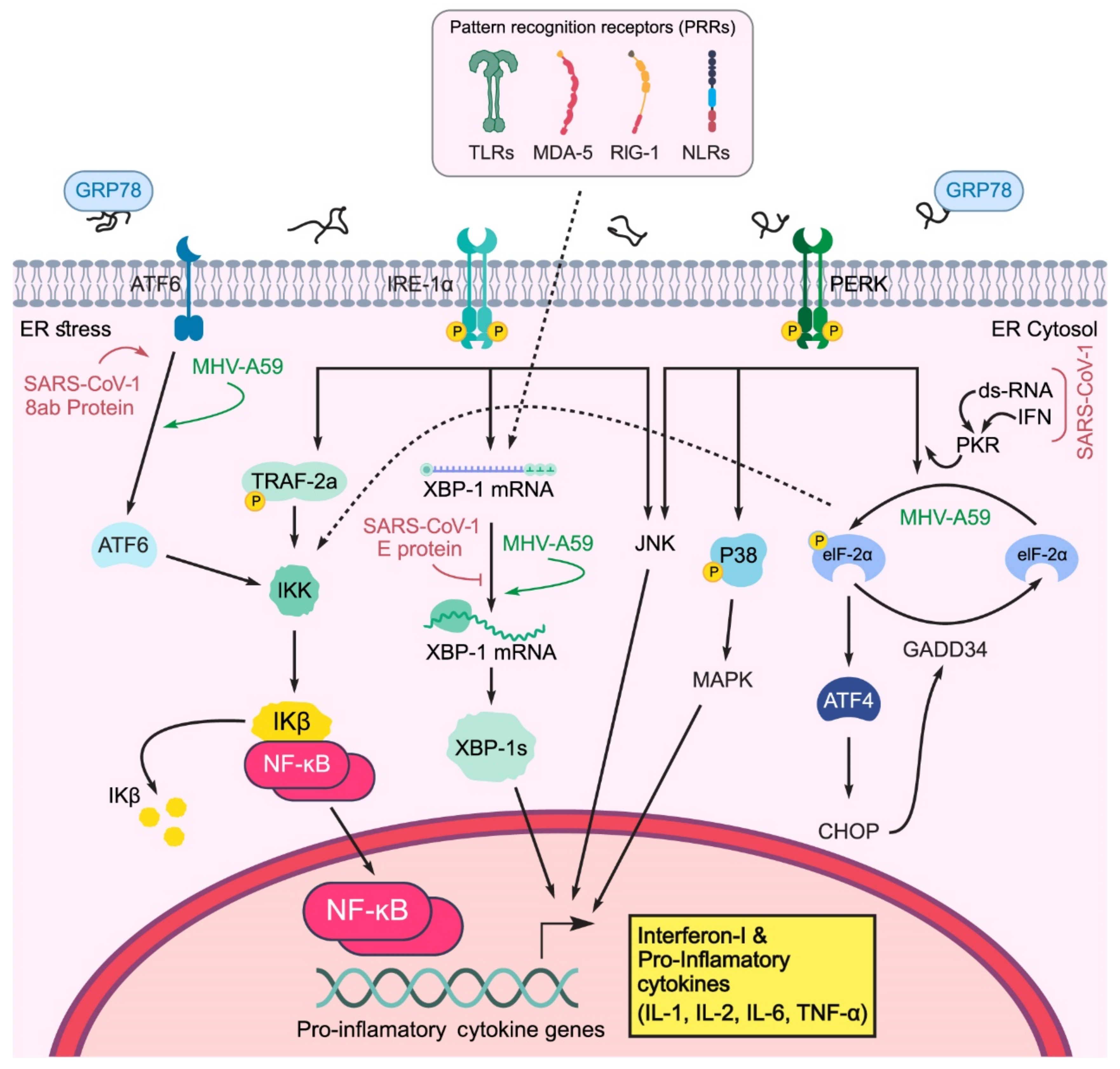
2.2. Effect of Coronavirus S and E Proteins on the UPR
2.3. Antiviral Targets
2.4. Effect of UPR on the Innate Immune Response
- PERK can inhibit the translation of IκBα by p-eIF2a, thereby activating NF-κB. Free accumulation of NF-κB causes NF-κB to activate and transfer to the nucleus, which leads to the expression of pro-inflammatory cytokines (IL-6, TNF-α, IL-1) [80];
- PERK can directly phosphorylate and activate JNK;
- PERK leads to p38 phosphorylation in the MAPK pathway.
3. Viral Infection and Autophagy
3.1. Coronavirus and Autophagy
3.2. Antivirals Targeting Autophagy
3.3. Role of Autophagy in the Innate Immune System
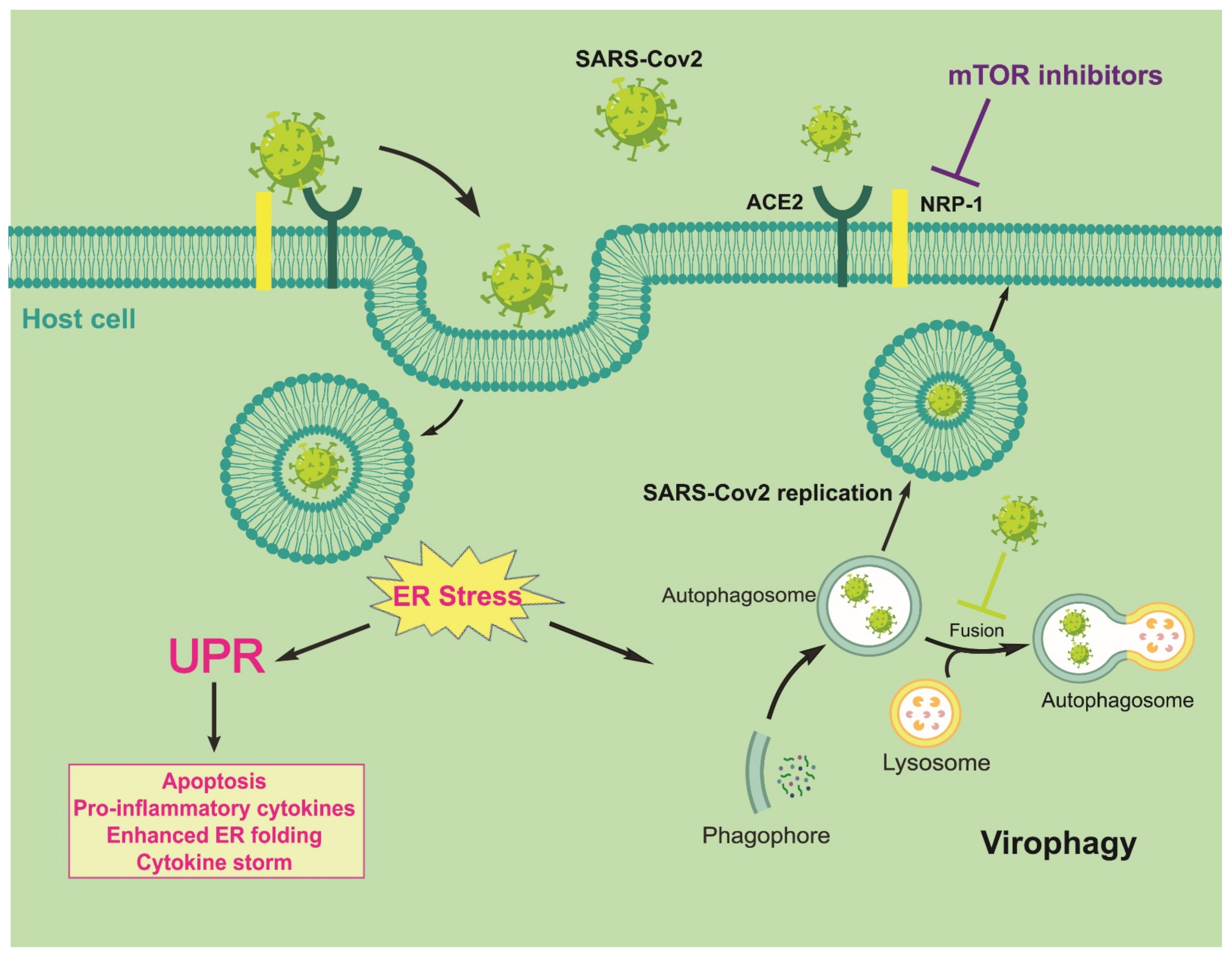
4. Discussion: Autophagy/UPR/mTOR/NRP1 Cross-Talk in SARS-CoV-2 Contagiousness as a Possible Target for Antiviral Activities and Innate/Acquired Immunity Modulation
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE2 | angiotensin-converting enzyme 2 |
| ACE2/Mas/Ang | ACE2-angiotensin-(1-7) and Mas receptor axis |
| ATF-6 | activation transcription factor-6 |
| Bcl-2 | B-cell lymphoma 2 |
| COVID-19 | coronavirus disease-19 |
| CHOP | C/EBP homologous protein |
| DAMP | danger-associated molecular patterns |
| DAPK | death-associated protein kinase |
| DCs | dendritic cells |
| DMVs | double membrane vesicles |
| DPP4 | dipeptidyl peptidase 4 |
| DDIT3 | DNA damage inducible transcript 3 |
| DR5 | death receptor 5 |
| DsRNA | double strand RNA |
| EGCG | Epigallocatechin-3-gallate |
| eIF2 α | eukaryotic initiation factor 2 α |
| ER | endoplasmic reticulum |
| EMCV | encephalomyocarditis virus |
| ERAD | ER-associated degradation |
| GADD153 | growth arrest- and DNA damage-inducible gene 153 |
| GBM | glioblastoma multiforme |
| HCV | hepatitis C virus |
| HECT | homologous to E6-AP carboxyl terminus |
| HSP | heat shock protein |
| ICTV | International Committee for Taxonomy of Viruses |
| IFNI | interferon I |
| IFN-β | interferon β |
| IRE-1 | inositol–requiring enzyme |
| IRGM | immunity-associated GTPase family M |
| ISG15 | interferon-stimulated gene 15 |
| JNK | c-Jun N-terminal kinases |
| KO | knocked out |
| MAPK | mitogen-activated protein kinase |
| MDA-5 | melanoma differentiation-associated protein 5 |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| mTOR | mechanistic target of the rapamycin complex 1 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NF-Y | nuclear transcription factor Y |
| NIC | niclosamide |
| NK | natural killer |
| NOD | nucleotidebinding oligomerization domain |
| NRP1 | neuropilin-1 |
| p58IPK | 58 kDa inhibitor protein kinase |
| PAMP | pathogen-associated molecular patterns |
| pDC | plasmacytoid DCs |
| PERK | Protein kinase R (PKR)-like ER kinase |
| PI3KC3 | phosphatidylinositol 3-kinase class III |
| PKR | protein kinase RNA-activated |
| PLP2-TM | membrane-associated papain-like protease |
| poly I:C | Polyinosinic:polycytidylic acid |
| PRRs | pattern recognition receptors |
| RIG-I | Retinoic acid-inducible Gene-I-like receptor |
| RIP | regulated intra-membrane proteolysis |
| SARS-CoV | severe acute respiratory syndrome coronavirus |
| SKP2 | S-phase kinase-associated protein 2 |
| SMURF1 | Smad ubiquitin regulatory factor 1 |
| TLR | Toll-like receptor |
| TRAF2 | TNF receptor-associated factor 2 |
| UPR | unfolded protein response |
| V-ATPase | vacuolar-type H+-ATPase |
| VAL | valinomycin |
| VSV | Vesicular stomatitis virus |
References
- Woolhouse, M.E.J.; Adair, K.; Brierley, L. RNA Viruses: A Case Study of the Biology of Emerging Infectious Diseases. One Health People Anim. Environ. 2014, 1, 81–97. [Google Scholar] [CrossRef]
- Khan, S.; Siddique, R.; Shereen, M.A.; Ali, A.; Liu, J.; Bai, Q.; Bashir, N.; Xue, M. Emergence of a Novel Coronavirus, Severe Acute Respiratory Syndrome Coronavirus 2: Biology and Therapeutic Options. J. Clin. Microbiol. 2020, 58, e00187-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Shcherbik, N.; Sawaya, B.E. Why do SARS-CoV-2 NSPs rush to the ER? J. Neurol. 2020, 267, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fecchi, K.; Anticoli, S.; Peruzzu, D.; Iessi, E.; Gagliardi, M.C.; Matarrese, P.; Ruggieri, A. Coronavirus Interplay With Lipid Rafts and Autophagy Unveils Promising Therapeutic Targets. Front. Microbiol. 2020, 11, 1821. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef] [PubMed]
- Payne, S. Family Coronaviridae. Viruses 2017, 149–158. [Google Scholar] [CrossRef]
- Satarker, S.; Nampoothiri, M. Structural Proteins in Severe Acute Respiratory Syndrome Coronavirus-2. Arch. Med Res. 2020, 51, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Qian, S.; Zhang, S.; Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 2020, 526, 135–140. [Google Scholar] [CrossRef]
- Sinha, N.; Balayla, G. Hydroxychloroquine and covid-19. Postgrad. Med. J. 2020, 96, 550. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Comish, P.; Kang, R. The hallmarks of COVID-19 disease. PLOS Pathog. 2020, 16, e1008536. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Chao, T.L.; Li, C.L.; Chiu, M.F.; Kao, H.C.; Wang, S.H.; Pang, Y.H.; Lin, C.H.; Tsai, Y.M.; Lee, W.H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020, 33, 108254. [Google Scholar] [CrossRef]
- Dastghaib, S.; Kumar, P.S.; Aftabi, S.; Damera, G.; Dalvand, A.; Sepanjnia, A.; Kiumarsi, M.; Aghanoori, M.-R.; Sohal, S.S.; Ande, S.R.; et al. Mechanisms Targeting the Unfolded Protein Response in Asthma. Am. J. Respir. Cell Mol. Biol. 2021, 64, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Jheng, J.-R.; Ho, J.-Y.; Horng, J.-T. ER stress, autophagy, and RNA viruses. Front. Microbiol. 2014, 5, 388. [Google Scholar] [CrossRef] [Green Version]
- Chadwick, S.R.; Lajoie, P. Endoplasmic Reticulum Stress Coping Mechanisms and Lifespan Regulation in Health and Diseases. Front. Cell Dev. Biol. 2019, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Casas, C. GRP78 at the Centre of the Stage in Cancer and Neuroprotection. Front. Neurosci. 2017, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Afshin, S. (Eds.) Endoplasmic Reticulum Stress in Health and Disease; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Doultsinos, D.; Avril, T.; Lhomond, S.; Dejeans, N.; Guédat, P.; Chevet, E. Control of the Unfolded Protein Response in Health and Disease. SLAS Discov. Adv. Life Sci. R&D 2017, 22, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Aghaei, M.; Dastghaib, S.; Aftabi, S.; Aghanoori, M.-R.; Alizadeh, J.; Mokarram, P.; Mehrbod, P.; Ashrafizadeh, M.; Zarrabi, A.; McAlinden, K.D.; et al. The ER Stress/UPR Axis in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Life 2020, 11, 1. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Talty, A.; Logue, S.E.; Mnich, K.; Gorman, A.M.; Samali, A. An Emerging Role for the Unfolded Protein Response in Pancreatic Cancer. Cancers 2021, 13, 261. [Google Scholar] [CrossRef]
- Rahmati, M.; Amanpour, S.; Kharman-Biz, A.; Moosavi, M.A. Endoplasmic Reticulum Stress as a Therapeutic Target in Cancer: A mini review. Basic Clin. Cancer Res. 2017, 9, 38–48. [Google Scholar]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signaling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic Reticulum Stress–Activated Cell Reprogramming in Oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Limia, C.; Sauzay, C.; Urra, H.; Hetz, C.; Chevet, E.; Avril, T. Emerging Roles of the Endoplasmic Reticulum Associated Unfolded Protein Response in Cancer Cell Migration and Invasion. Cancers 2019, 11, 631. [Google Scholar] [CrossRef] [Green Version]
- Emami, A.; Shojaei, S.; Rosa, S.C.D.S.; Aghaei, M.; Samiei, E.; Vosoughi, A.R.; Kalantari, F.; Kawalec, P.; Thliveris, J.; Sharma, P.; et al. Mechanisms of simvastatin myotoxicity: The role of autophagy flux inhibition. Eur. J. Pharmacol. 2019, 862, 172616. [Google Scholar] [CrossRef] [PubMed]
- Dastghaib, S.; Shojaei, S.; Mostafavi-Pour, Z.; Sharma, P.; Patterson, J.; Samali, A.; Mokarram, P.; Ghavami, S. Simvastatin Induces Unfolded Protein Response and Enhances Temozolomide-Induced Cell Death in Glioblastoma Cells. Cells 2020, 9, 2339. [Google Scholar] [CrossRef]
- Mohammad Fazlul, K.; Hyung-Ryong, K.; Han-Jung, C. Endoplasmic Reticulum Stress and Autophagy. In Endoplasmic Reticulum; Català, A., Ed.; IntechOpen: London, UK, 2019. [Google Scholar]
- Zhang, C.; Syed, T.W.; Liu, R.; Yu, J. Role of Endoplasmic Reticulum Stress, Autophagy, and Inflammation in Cardiovascular Disease. Front. Cardiovasc. Med. 2017, 4, 29. [Google Scholar] [CrossRef]
- Margariti, A.; Li, H.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.; Zampetaki, A.; Zhang, Z.; et al. XBP1 mRNA Splicing Triggers an Autophagic Response in Endothelial Cells through BECLIN-1 Transcriptional Activation. J. Biol. Chem. 2013, 288, 859–872. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Kanekura, K.; Levine, T.P.; Kohno, K.; Olkkonen, V.M.; Aiso, S.; Matsuoka, M. ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J. Neurochem. 2009, 108, 973–985. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, H.W.; Charnock-Jones, D.S.; Burton, G.J. Regulation of AKT Phosphorylation at Ser473 and Thr308 by Endoplasmic Reticulum Stress Modulates Substrate Specificity in a Severity Dependent Manner. PLoS ONE 2011, 6, e17894. [Google Scholar] [CrossRef] [PubMed]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Łos, M.J.; Clementi, E.; Ghavami, S. Endoplasmic reticulum as a potential therapeutic target for covid-19 infection management? Eur. J. Pharmacol. 2020, 882, 173288. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, K.; Mao, B.; Liu, S.; Trilling, M.; Huang, A.; Lu, M.; Lin, Y. The interplay between emerging human coronavirus infections and autophagy. Emerg. Microbes Infect. 2021, 10, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020, 19, 155–170. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Coronavirus infection, ER stress, apoptosis and innate immunity. Front. Microbiol. 2014, 5, 296. [Google Scholar] [CrossRef] [Green Version]
- Ha, D.P.; Van Krieken, R.; Carlos, A.J.; Lee, A.S. The stress-inducible molecular chaperone GRP78 as potential therapeutic target for coronavirus infection. J. Infect. 2020, 81, 452–482. [Google Scholar] [CrossRef]
- Li, S.; Kong, L.; Yu, X. The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit. Rev. Microbiol. 2013, 41, 150–164. [Google Scholar] [CrossRef]
- Krähling, V.; Stein, D.A.; Spiegel, M.; Weber, F.; Mühlberger, E. Severe Acute Respiratory Syndrome Coronavirus Triggers Apoptosis via Protein Kinase R but Is Resistant to Its Antiviral Activity. J. Virol. 2008, 83, 2298–2309. [Google Scholar] [CrossRef] [Green Version]
- Echavarría-Consuegra, L.; Cook, G.M.; Busnadiego, I.; Lefèvre, C.; Keep, S.; Brown, K.; Doyle, N.; Dowgier, G.; Franaszek, K.; Moore, N.A.; et al. Manipulation of the unfolded protein response: A pharmacological strategy against coronavirus infection. bioRxiv 2021, 292979. [Google Scholar] [CrossRef] [Green Version]
- Versteeg, G.; Van De Nes, P.S.; Bredenbeek, P.J.; Spaan, W.J.M. The Coronavirus Spike Protein Induces Endoplasmic Reticulum Stress and Upregulation of Intracellular Chemokine mRNA Concentrations. J. Virol. 2007, 81, 10981–10990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechill, J.; Chen, Z.; Brewer, J.W.; Baker, S.C. Coronavirus Infection Modulates the Unfolded Protein Response and Mediates Sustained Translational Repression. J. Virol. 2008, 82, 4492–4501. [Google Scholar] [CrossRef] [Green Version]
- De Diego, M.L.; Nieto-Torres, J.L.; Guardeno, J.M.J.; Regla-Nava, J.A.; Álvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Regulates Cell Stress Response and Apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar] [CrossRef] [Green Version]
- Sung, S.-C.; Chao, C.-Y.; Jeng, K.-S.; Yang, J.-Y.; Lai, M.M. The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology 2009, 387, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liao, Y.; Liu, D.X. Regulation of Stress Responses and Translational Control by Coronavirus. Viruses 2016, 8, 184. [Google Scholar] [CrossRef]
- Shah, S.Z.A.; Zhao, D.; Hussain, T.; Yang, L. The Role of Unfolded Protein Response and Mitogen-Activated Protein Kinase Signaling in Neurodegenerative Diseases with Special Focus on Prion Diseases. Front. Aging Neurosci. 2017, 9, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Conza, G.; Ho, P.-C. ER Stress Responses: An Emerging Modulator for Innate Immunity. Cells 2020, 9, 695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaei, S.; Suresh, M.; Klionsky, D.J.; Labouta, H.I.; Ghavami, S. Autophagy and SARS-CoV-2 infection: A possible smart targeting of the autophagy pathway. Virulence 2020, 11, 805–810. [Google Scholar] [CrossRef]
- Boga, J.A.; Coto-Montes, A. ER stress and autophagy induced by SARS-CoV-2: The targets for melatonin treatment. Melatonin Res. 2020, 3, 346–361. [Google Scholar] [CrossRef]
- Chu, H.; Chan, C.M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.H.; Sze, K.H.; Yang, D.; Shuai, H.; et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.-L.; Chan, C.-P.; Kok, K.-H.; Woo, P.C.-Y.; Jin, D.-Y. Comparative analysis of the activation of unfolded protein response by spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus HKU1. Cell Biosci. 2014, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, N.; Anathy, V. Pathological consequences of the unfolded protein response and downstream protein disulphide isomerases in pulmonary viral infection and disease. J. Biochem. 2019, 167, 173–184. [Google Scholar] [CrossRef]
- Kushwaha, R.S.; Nayak, P.K. A review on pharmacological targets for treatment of COVID-19 infection. J. Adv. Sci. Res. 2020, 11, 1–15. [Google Scholar]
- Chan, C.-P.; Siu, K.-L.; Chin, K.-T.; Yuen, K.-Y.; Zheng, B.; Jin, D.-Y. Modulation of the Unfolded Protein Response by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during Coronavirus Infectious Bronchitis Virus Infection Modulates Apoptosis by Restricting Activation of the Extracellular Signal-Regulated Kinase Pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [Green Version]
- Robert, J. Evolution of heat shock protein and immunity. Dev. Comp. Immunol. 2003, 27, 449–464. [Google Scholar] [CrossRef]
- Rhimi, P.; Aghasadeghi, M.R. Prospective on Different Approaches for Vaccine Development against COVID-19: Past Lessons and Future Challenges. Vaccine Res. 2019, 6, 14–17. [Google Scholar] [CrossRef]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The SARS Coronavirus 3a Protein Causes Endoplasmic Reticulum Stress and Induces Ligand-Independent Downregulation of the Type 1 Interferon Receptor. PLoS ONE 2009, 4, e8342. [Google Scholar] [CrossRef]
- Rabouw, H.H.; Langereis, M.A.; Knaap, R.C.M.; Dalebout, T.J.; Canton, J.; Sola, I.; Enjuanes, L.; Bredenbeek, P.J.; Kikkert, M.; De Groot, R.J.; et al. Middle East Respiratory Coronavirus Accessory Protein 4a Inhibits PKR-Mediated Antiviral Stress Responses. PLoS Pathog. 2016, 12, e1005982. [Google Scholar] [CrossRef] [PubMed]
- Bradley, C.A. Emerging role for the unfolded protein response. Nat. Rev. Urol. 2019, 16, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Le Reste, P.J.; Pineau, R.; Voutetakis, K.; Samal, J.; Jégou, G.; Lhomond, S.; Gorman, A.M.; Samali, A.; Patterson, J.B.; Zeng, Q.; et al. Local intracerebral inhibition of IRE1 by MKC8866 sensitizes glioblastoma to irradiation/chemotherapy in vivo. Cancer Lett. 2020, 494, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Czinn, S.J.; Reiter, R.J.; Blanchard, T.G. Crosstalk between endoplasmic reticulum stress and anti-viral activities: A novel therapeutic target for COVID-19. Life Sci. 2020, 255, 117842. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Korkmaz, A.; Paredes, S.D.; Manchester, L.C.; Tan, D.-X. Melatonin reduces oxidative/nitrosative stress due to drugs, toxins, metals, and herbicides. Neuro Endocrinol. Lett. 2008, 29, 609–613. [Google Scholar]
- Murakami, Y.; Ishikawa, K.; Nakao, S.; Sonoda, K.-H. Innate immune response in retinal homeostasis and inflammatory disorders. Prog. Retin. Eye Res. 2020, 74, 100778. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Ambesi, A.; McKeown-Longo, P.J. Role of TLR4 Receptor Complex in the Regulation of the Innate Immune Response by Fibronectin. Cells 2020, 9, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malathi, K.; Dong, B.; Gale, M.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Mol. Cell Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Ma, S.; Yin, X.; Yi, P.; Liu, J. An integrated PKD1-dependent signaling network amplifies IRE1 prosurvival signaling. J. Biol. Chem. 2019, 294, 11119–11130. [Google Scholar] [CrossRef]
- Martinon, F.; Chen, X.; Lee, A.-H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Yu, X.; Wang, H.; Zuo, D.; Guo, C.; Yi, H.; Tirosh, B.; Subjeck, J.R.; Qiu, X.; Wang, X.-Y. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur. J. Immunol. 2011, 41, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007, 204, 2267–2275. [Google Scholar] [CrossRef]
- Wu, S.; Tan, M.; Hu, Y.; Wang, J.-L.; Scheuner, D.; Kaufman, R.J. Ultraviolet light activates NFκB through translational inhibition of IκBα synthesis. J. Biol. Chem. 2004, 279, 34898–34902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavarino, G.; Cláudio, N.; Couderc, T.; Dalet, A.; Judith, D.; Camosseto, V.; Schmidt, E.K.; Wenger, T.; Lecuit, M.; Gatti, E. Induction of GADD34 is necessary for dsRNA-dependent interferon-β production and participates in the control of Chikungunya virus infection. PLoS Pathog. 2012, 8, e1002708. [Google Scholar] [CrossRef] [Green Version]
- Clavarino, G.; Cláudio, N.; Dalet, A.; Terawaki, S.; Couderc, T.; Chasson, L.; Ceppi, M.; Schmidt, E.K.; Wenger, T.; Lecuit, M.; et al. Protein phosphatase 1 subunit Ppp1r15a/GADD34 regulates cytokine production in polyinosinic:polycytidylic acid-stimulated dendritic cells. Proc. Natl. Acad. Sci. USA 2012, 109, 3006–3011. [Google Scholar] [CrossRef] [Green Version]
- Goodall, J.C.; Wu, C.; Zhang, Y.; McNeill, L.; Ellis, L.; Saudek, V.; Gaston, J.S.H. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17698–17703. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Barrera, N.; Pavicic, P.G., Jr.; Zhao, C.; Freeman, M.; Min, B.; Hamilton, T. cEBP Homologous protein expression in macrophages regulates the magnitude and duration of IL-6 expression and dextran sodium sulfate colitis. J. Interf. Cytok. Res. 2015, 35, 785–794. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.; Yue, S.; Fu, Y.; Zhu, J.; Wang, X.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Zhai, Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am. J. Transpl. 2014, 14, 1552–1561. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, R.L.; MacKenzie, J.M. ATF6 Signaling Is Required for Efficient West Nile Virus Replication by Promoting Cell Survival and Inhibition of Innate Immune Responses. J. Virol. 2013, 87, 2206–2214. [Google Scholar] [CrossRef] [Green Version]
- Carletti, T.; Zakaria, M.K.; Faoro, V.; Reale, L.; Kazungu, Y.; Licastro, D.; Marcello, A. Viral priming of cell intrinsic innate antiviral signaling by the unfolded protein response. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Wang, X.; Huang, M.; Tam, J.P.; Liu, D.X. Regulation of the p38 mitogen-activated protein kinase and dual-specificity phosphatase 1 feedback loop modulates the induction of interleukin 6 and 8 in cells infected with coronavirus infectious bronchitis virus. Virology 2011, 420, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoji-Kawata, S.; Levine, B. Autophagy, antiviral immunity, and viral countermeasures. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1793, 1478–1484. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.; Gu, M.J.; Kim, C.G.; Kye, Y.C.; Lim, Y.; Lee, J.E.; Park, B.-C.; Chu, H.; Han, S.H.; Yun, C.-H. Rapamycin-induced autophagy restricts porcine epidemic diarrhea virus infectivity in porcine intestinal epithelial cells. Antivir. Res. 2017, 146, 86–95. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, M.; Zhang, X.; Tan, X.; Guo, H.; Zeng, W.; Yan, G.; Memon, A.M.; Li, Z.; Zhu, Y.; et al. Porcine Epidemic Diarrhea Virus Induces Autophagy to Benefit Its Replication. Viruses 2017, 9, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Yu, H.; Gu, W.; Luo, X.; Li, R.; Zhang, J.; Xu, Y.; Yang, L.; Shen, N.; Feng, L.; et al. Autophagy Negatively Regulates Transmissible Gastroenteritis Virus Replication. Sci. Rep. 2016, 6, 23864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Levine, B. Autophagy and Viruses: Adversaries or Allies? J. Innate Immun. 2013, 5, 480–493. [Google Scholar] [CrossRef]
- Orvedahl, A.; Sumpter, R.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.; Ackermann, K.; Stuart, M.; Wex, C.; Protzer, U.; Schätzl, H.M.; Gilch, S. Severe Acute Respiratory Syndrome Coronavirus Replication Is Severely Impaired by MG132 due to Proteasome-Independent Inhibition of M-Calpain. J. Virol. 2012, 86, 10112–10122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.; Denison, M.R.; Virgin, I.; Herbert, W. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007, 3, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Reggiori, F. ATG proteins: Are we always looking at autophagy? Autophagy 2016, 12, 2502–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yordy, B.; Tal, M.; Hayashi, K.; Arojo, O.; Iwasaki, A. Autophagy and selective deployment of Atg proteins in antiviral defense. Int. Immunol. 2013, 25, 1–10. [Google Scholar] [CrossRef]
- Tal, M.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef] [Green Version]
- Hanning, J.; Saini, H.K.; Murray, M.J.; Caffarel, M.M.; Van Dongen, S.; Ward, D.; Barker, E.M.; Scarpini, C.G.; Groves, I.J.; Stanley, M.; et al. Depletion of HPV16 early genes induces autophagy and senescence in a cervical carcinogenesis model, regardless of viral physical state. J. Pathol. 2013, 231, 354–366. [Google Scholar] [CrossRef]
- Fliss, P.M.; Jowers, T.P.; Brinkmann, M.M.; Holstermann, B.; Mack, C.; Dickinson, P.; Hohenberg, H.; Ghazal, P.; Brune, W. Viral Mediated Redirection of NEMO/IKKγ to Autophagosomes Curtails the Inflammatory Cascade. PLoS Pathog. 2012, 8, e1002517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichinose, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes through intracellular M2 channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef]
- Yeganeh, B.; Ghavami, S.; Rahim, N.; Klonisch, T.; Halayko, A.; Coombs, K. Autophagy activation is required for influenza A virus-induced apoptosis and replication. Biochim. Biophys. Acta BBA Bioenerg. 2018, 1865, 364–378. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Lin, X.; Ren, C.; Zhao, J.; Wang, F.; Gao, X.; Xiao, R.; Zhao, L.; Chen, H.; et al. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy 2019, 15, 1163–1181. [Google Scholar] [CrossRef]
- Yeganeh, B.; Moghadam, A.R.; Alizadeh, J.; Wiechec, E.; Alavian, S.M.; Hashemi, M.; Geramizadeh, B.; Samali, A.; Lankarani, K.B.; Post, M.; et al. Hepatitis B and C virus-induced hepatitis: Apoptosis, autophagy, and unfolded protein response. World J. Gastroenterol. 2015, 21, 13225–13239. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.M.; Gong, B.; Chan, T.; Davey, R.A.; Soong, L.; Kolokoltsov, A.A.; Sun, J. Differential, Type I Interferon-Mediated Autophagic Trafficking of Hepatitis C Virus Proteins in Mouse Liver. Gastroenterology 2011, 141, 674–685.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Ou, J.-H.J. Hepatitis C virus and autophagy. Biol. Chem. 2015, 396, 1215–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Da, L.; Mao, Y.; Li, Y.; Li, D.; Xu, Z.; Li, F.; Wang, Y.; Tiollais, P.; Li, T.; et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology 2008, 49, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Liu, Y.; Zeng, X.; Wei, M.; Wu, S.; Xiong, Q.; Song, F.; Yuan, X.; Xiao, Y.; et al. Hepatitis B Virus Induces Autophagy to Promote its Replication by the Axis of miR-192-3p-XIAP Through NF kappa B Signaling. Hepatology 2019, 69, 974–992. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Tian, Y.; Chen, W.-L.; Ann, D.K.; Yen, T.-S.B.; Ou, J.-H.J. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Chowdhury, J.B.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C Virus Upregulates Beclin1 for Induction of Autophagy and Activates mTOR Signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef] [Green Version]
- Su, W.-C.; Chao, T.-C.; Huang, Y.-L.; Weng, S.-C.; Jeng, K.-S.; Lai, M.M.C. Rab5 and Class III Phosphoinositide 3-Kinase Vps34 Are Involved in Hepatitis C Virus NS4B-Induced Autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grégoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM Is a Common Target of RNA Viruses that Subvert the Autophagy Network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef]
- Liu, B.; Fang, M.; Hu, Y.; Huang, B.; Li, N.; Chang, C.; Huang, R.; Xu, X.; Yang, Z.; Chen, Z.; et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 2013, 10, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tian, Y.; Ou, J.-H.J. HCV Induces the Expression of Rubicon and UVRAG to Temporally Regulate the Maturation of Autophagosomes and Viral Replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Kang, R.; Wang, J.; Luo, G.; Yang, W.; Zhao, Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013, 9, 175–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L. Autophagy in hepatitis B or C virus infection: An incubator and a potential therapeutic target. Life Sci. 2020, 242, 117206. [Google Scholar] [CrossRef]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Zhu, W.; Cao, S.; Chen, R.; Jin, H.; Liu, Y.; Wang, S.; Wang, W.; Xiao, G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE 2013, 8, e52909. [Google Scholar]
- Tu, Y.-C.; Yu, C.-Y.; Liang, J.-J.; Lin, E.; Liao, C.-L.; Lin, Y.-L. Blocking Double-Stranded RNA-Activated Protein Kinase PKR by Japanese Encephalitis Virus Nonstructural Protein 2A. J. Virol. 2012, 86, 10347–10358. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.D.; Torres, V.J.; Cadwell, K. Autophagy and microbial pathogenesis. Cell Death Differ. 2020, 27, 872–886. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Gutierrez, D.; Bauer, M.A.; Zimmermann, A.; Kainz, K.; Hofer, S.J.; Kroemer, G.; Madeo, F. Digesting the crisis: Autophagy and coronaviruses. Microb. Cell 2020, 7, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; McGrath, M.E.; Hu, Z.; Ariannejad, S.; Weston, S.; Frieman, M.; Jackson, W.T. Coronavirus interactions with the cellular autophagy machinery. Autophagy 2020, 16, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, K.; Xing, Y.; Tu, J.; Yang, X.; Zhao, Q.; Li, K.; Chen, Z. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell 2014, 5, 912–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijaljica, D.; Klionsky, D.J. Autophagy/virophagy: A “disposal strategy” to combat COVID-19. Autophagy 2020, 16, 2271–2272. [Google Scholar] [CrossRef]
- Yeganeh, B.; Ghavami, S.; Kroeker, A.L.; Mahood, T.H.; Stelmack, G.L.; Klonisch, T.; Coombs, K.M.; Halayko, A.J. Suppression of influenza A virus replication in human lung epithelial cells by noncytotoxic concentrations bafilomycin A1. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L270–L286. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Xue, J.; Yang, Z.; Shi, Y.; Shi, Y.; Lou, G.; Wu, S.; Qi, J.; Liu, W.; et al. MicroRNA-141 Targets Sirt1 and Inhibits Autophagy to Reduce HBV Replication. Cell. Physiol. Biochem. 2017, 41, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Kunanopparat, A.; Hirankarn, N.; Kittigul, C.; Tangkijvanich, P.; Kimkong, I. Autophagy machinery impaired interferon signalling pathways to benefit hepatitis B virus replication. Asian Pac. J. Allergy Immunol. 2016, 34, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Hu, J.; Shu, W.; Gao, B.; Xiong, S. Epigallocatechin-3-gallate opposes HBV-induced incomplete autophagy by enhancing lysosomal acidification, which is unfavorable for HBV replication. Cell Death Dis. 2015, 6, e1770. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Yuan, K.; Zhou, L.; Wang, K.; Chen, H.-N.; Lei, Y.; Lan, J.; Pu, Q.; Gao, W.; Zhang, L.; et al. PRKAA/AMPK restricts HBV replication through promotion of autophagic degradation. Autophagy 2016, 12, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Elgner, F.; Jiang, B.; Himmelsbach, K.; Medvedev, R.; Ploen, D.; Hildt, E. The autophagosomal SNARE Pprotein syntaxin 17 is an essential factor for the hepatitis C virus life cycle. J. Virol. 2016, 90, 5989–6000. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Kim, M.-J.; Sung, P.S.; Bae, Y.C.; Shin, E.-C.; Yoo, J.-Y. Interferon-inducible protein SCOTIN interferes with HCV replication through the autolysosomal degradation of NS5A. Nat. Commun. 2016, 7, 10631. [Google Scholar] [CrossRef] [Green Version]
- Abubakar, A.R.; Sani, I.H.; Godman, B.; Kumar, S.; Islam, S.; Jahan, I.; Haque, M. Systematic Review on the Therapeutic Options for COVID-19: Clinical Evidence of Drug Efficacy and Implications. Infect. Drug Resist. 2020, 13, 4673–4695. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Wang, H.; Yang, Y.; Chen, Z.-S.; Zou, C.; Zhang, J. Chloroquine against malaria, cancers and viral diseases. Drug Discov. Today 2020, 25, 2012–2022. [Google Scholar] [CrossRef]
- Endy, T.P.; Keiser, P.B.; Cibula, D.; Abbott, M.; Ware, L.; Thomas, S.J.; Polhemus, M.E. Effect of Antimalarial Drugs on the Immune Response to Intramuscular Rabies Vaccination Using a Postexposure Prophylaxis Regimen. J. Infect. Dis. 2019, 221, 927–933. [Google Scholar] [CrossRef]
- Oscanoa, T.J.; Romero-Ortuno, R.; Carvajal, A.; Savarino, A. A pharmacological perspective of chloroquine in SARS-CoV-2 infection: An old drug for the fight against a new coronavirus? Int. J. Antimicrob. Agents 2020, 56, 106078. [Google Scholar] [CrossRef]
- Ho, T.-C.; Wang, Y.-H.; Chen, Y.-L.; Tsai, W.-C.; Lee, C.-H.; Chuang, K.-P.; Chen, Y.-M.; Yuan, C.-H.; Ho, S.-Y.; Yang, M.-H.; et al. Chloroquine and Hydroxychloroquine: Efficacy in the Treatment of the COVID-19. Pathogens 2021, 10, 217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Li, L.; Feng, Z.; Wan, S.; Huang, P.; Sun, X.; Wen, F.; Huang, X.; Ning, G.; Wang, W. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motaghinejad, M.; Mesrabadi, M.A.; Gholami, M.; Safari, S. Possible Involvement of Autophagy or Apoptosis Dysregulation in Infection with COVID-19 Virus as the Main Cause of Mortality: Hypothetical Function of ACE-2/Mas / Ang (1–7) Signaling Pathway and Proposal of a Post-infection Treatment Strategy. Crit. Comments Biomed. 2020, 1, 100–124. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Mehrbod, P.; Hair-Bejo, M.; Ibrahim, T.A.T.; Omar, A.R.; El Zowalaty, M.; Ajdari, Z.; Ideris, A. Simvastatin modulates cellular components in influenza A virus-infected cells. Int. J. Mol. Med. 2014, 34, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Zhang, P.; Li, N.; Kiang, K.M.Y.; Cheng, S.Y.; Wong, V.K.W.; Leung, G.K.-K. Lovastatin Enhances Cytotoxicity of Temozolomide via Impairing Autophagic Flux in Glioblastoma Cells. BioMed Res. Int. 2019, 2019, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Drożdżal, S.; Rosik, J.; Lechowicz, K.; Machaj, F.; Kotfis, K.; Ghavami, S.; Łos, M.J. FDA approved drugs with pharmacotherapeutic potential for SARS-CoV-2 (COVID-19) therapy. Drug Resist. Updates 2020, 53, 100719. [Google Scholar] [CrossRef]
- Panda, P.K.; Fahrner, A.; Vats, S.; Seranova, E.; Sharma, V.; Chipara, M.; Desai, P.; Torresi, J.; Rosenstock, T.; Kumar, D.; et al. Chemical Screening Approaches Enabling Drug Discovery of Autophagy Modulators for Biomedical Applications in Human Diseases. Front. Cell Dev. Biol. 2019, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.; Mutawe, M.M.; Sharma, P.; Yeganeh, B.; McNeill, K.D.; Klonisch, T.; Unruh, H.; Kashani, H.H.; Schaafsma, D.; Los, M.; et al. Mevalonate Cascade Regulation of Airway Mesenchymal Cell Autophagy and Apoptosis: A Dual Role for p53. PLoS ONE 2011, 6, e16523. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Sharma, P.; Yeganeh, B.; Ojo, O.O.; Jha, A.; Mutawe, M.M.; Kashani, H.H.; Los, M.J.; Klonisch, T.; Unruh, H.; et al. Airway mesenchymal cell death by mevalonate cascade inhibition: Integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim. Biophys. Acta BBA Bioenerg. 2014, 1843, 1259–1271. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Cui, R.; Ding, T.; Li, X.; Peng, J.; Xu, W.; Han, F.; Guo, X. Simvastatin alleviates airway inflammation and remodelling through up-regulation of autophagy in mouse models of asthma. Respirology 2017, 22, 533–541. [Google Scholar] [CrossRef]
- Peymani, P.; Dehesh, T.; Aligolighasemabadi, F.; Sadeghdoust, M.; Kotfis, K.; Ahmadi, M.; Mehrbod, P.; Iranpour, P.; Dastghaib, S.; Nasimian, A.; et al. Statins in patients with COVID-19: A retrospective cohort study in Iranian COVID-19 patients. Transl. Med. Commun. 2021, 6, 1–14. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Liu, J.; Liang, H.; Chen, C.; Wang, X.; Qu, F.; Wang, H.; Yang, K.; Wang, Q.; Zhao, N.; Meng, J.; et al. Ivermectin induces autophagy-mediated cell death through the AKT/mTOR signaling pathway in glioma cells. Biosci. Rep. 2019, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- García-Pérez, B.E.; González-Rojas, J.A.; Salazar, M.I.; Torres-Torres, C.; Castrejón-Jiménez, N.S. Taming the Autophagy as a Strategy for Treating COVID-19. Cells 2020, 9, 2679. [Google Scholar] [CrossRef]
- Fakher, S.; Peymani, P.; Ghavami, S.; Mokarram, P. The Role of Autophagy in Respiratory Complications of COVID-19. Shiraz E-Med. J. 2020, 21, 102967. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0 s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cao, H.; Li, J.; Wang, B.; Zhang, P.; Zhang, X.D.; Liu, Z.; Yuan, H.; Zhan, Z. Autophagy induced by DAMPs facilitates the inflammation response in lungs undergoing ischemia-reperfusion injury through promoting TRAF6 ubiquitination. Cell Death Differ. 2017, 24, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.-S.; Shenderov, K.; Huang, N.-N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Nazinitsky, A.; Rosenthal, K.S. Cytokine storms: Systemic disasters of infectious diseases. Infect. Dis. Clin. Pract. 2010, 18, 188–192. [Google Scholar] [CrossRef]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.-M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.-T.; Meng, X.-J.; Li, L.-L.; Liu, Y.; Li, W.-F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.; Shin, D.-M.; Ramakrishna, L.; Goussetis, D.J.; Platanias, L.C.; Xiong, H.; Morse, H.C., III; Ozato, K. IRF8 directs stress-induced autophagy in macrophages and promotes clearance of Listeria monocytogenes. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Trouillet-Assant, S.; Viel, S.; Gaymard, A.; Pons, S.; Richard, J.-C.; Perret, M.; Villard, M.; Brengel-Pesce, K.; Lina, B.; Mezidi, M.; et al. Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 2020, 146, 206–208.e2. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.-E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Pang, H.-B.; Braun, G.B.; Friman, T.; Aza-Blanc, P.; Ruidiaz, M.E.; Sugahara, K.N.; Teesalu, T.; Ruoslahti, E. An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, C.; Lechien, J.R.; Saussez, S. More that ACE2? NRP1 may play a central role in the underlying pathophysiological mechanism of olfactory dysfunction in COVID-19 and its association with enhanced survival. Med. Hypotheses 2021, 146, 110406. [Google Scholar] [CrossRef]
- Roy, S.; Bag, A.K.; Singh, R.K.; Talmadge, J.E.; Batra, S.K.; Datta, K. Multifaceted Role of Neuropilins in the Immune System: Potential Targets for Immunotherapy. Front. Immunol. 2017, 8, 1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.R.; Berrien-Elliott, M.; Yuan, J.; Hsueh, E.C.; Teague, R.M. Neuropilin-1 Expression Is Induced on Tolerant Self-Reactive CD8+ T Cells but Is Dispensable for the Tolerant Phenotype. PLoS ONE 2014, 9, e110707. [Google Scholar] [CrossRef] [Green Version]
- Grage-Griebenow, E.; Löseke, S.; Kauth, M.; Gehlhar, K.; Zawatzky, R.; Bufe, A. Anti-BDCA-4 (neuropilin-1) antibody can suppress virus-induced IFN-alpha production of plasmacytoid dendritic cells. Immunol. Cell Biol. 2007, 85, 383–390. [Google Scholar] [CrossRef]
- Seadawy, M.; Shamel, M.; Ahmed, A.; Zekri, A.R.N. In Silico Docking for Inhibition Neuropilin-1 (SARS-CoV-2 Receptor) by Some Natural and Approved Drugs. Available online: https://ssrn.com/abstract=3735823 (accessed on 23 October 2020).
- Zeng, Q.; Sun, X.; Xiao, L.; Xie, Z.; Bettini, M.; Deng, T. A Unique Population: Adipose-Resident Regulatory T Cells. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Mönkemüller, K.; Fry, L.; Rickes, S. COVID-19, coronavirus, SARS-CoV-2 and the small bowel. Rev. Esp. Enferm. Dig. 2020, 112, 383–388. [Google Scholar]
- de Oliveira, A.P.; Lopes, A.L.; Pacheco, G.; Nolêto, I.R.; Nicolau, L.A.; Medeiros, J.V. Premises among SARS-CoV-2, dysbiosis and diarrhea: Walking through the ACE2/mTOR/autophagy route. Med. Hypotheses 2020, 144, 110243. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Kuo, S.-H.; Lin, C.-Y.; Fu, P.-J.; Lin, Y.-S.; Yeh, T.-M.; Liu, H.-S. Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nabirotchkin, S.; Peluffo, A.E.; Bouaziz, J.; Cohen, D. Focusing on the unfolded protein response and autophagy related pathways to reposition common approved drugs against COVID-19. Preprints 2020, 2020030302. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CoV Strain | Host System | Main Finding | Author & Year | ||
|---|---|---|---|---|---|
| MERS-CoV and bat coronavirus HKU9 | Cells | A549 | MERS-CoV and bat coronavirus HKU9 can use GRP78 as an extra target for attachment, which can exemplify the evolutionary adaptation of animal coronaviruses to human | Chu H, 2018 [56] | |
| AD293 | |||||
| HeLa | |||||
| Huh7 | |||||
| Caco2 | |||||
| VeroE6 | |||||
| Human | Primary human monocyte-derived macrophages | ||||
| Primary T cells were isolated from PBMCs | |||||
| SARS-1 | 293/ACE2 cells | Infection with SARS-CoV leads to the activation of PKR/PERK and triggers apoptosis independent of eIF2α phosphorylation | Krähling V, 2009 [44] | ||
| SARS-1 | Cells | COS-1 | The SARS-CoV 3a protein induces ER stress responses and degrades type 1 interferon receptor, which may reduce antiviral immune responses | Minakshi R, 2009 [65] | |
| Vero | |||||
| Huh7 | |||||
| MHV-A59 | 17Cl-1 cells | All arms of UPR are up-regulated with MHV-A59 infection | Cook GM, 2019 [45] | ||
| MERS-CoV | Cells | HeLa-R19 | The MERS-CoV 4a protein, as an effective stress antagonist, suppresses PKR-mediated stress responses that may promote apoptosis | Rabouw HH, 2016 [66] | |
| Huh7 | |||||
| BHK-21 | |||||
| Vero | |||||
| SARS-1, MHV-A59 | L-ACE2 cells | In CoV-infected cells, ER stress responses are induced by up-regulating XBP-1 mRNA and Herpud1, resulting in cytokine overexpression | Versteeg GA, 2007 [46] | ||
| MHV-A59 | Cells | DBT | Coronavirus infection by inducing UPR, suppresses the host cell protein synthesis to accelerate the viral protein translation | Bechill J, 2008 [47] | |
| HeLa-MHVR | |||||
| SARS-1 | Cells | HeLa | The SARS-1 8ab protein stimulates UPR to enable viral protein folding and processing by attaching to the luminal domain of ATF6 | Sung S-C, 2009 [49] | |
| Vero E6 | |||||
| SARS-1 | Cells | Animal | Vero E6 | The envelope protein of SARS-1 enhances ER stress responses and pro-inflammatory cytokine expression | DeDiego ML, 2011 [48] |
| MA-104 | |||||
| FRhK-4 | |||||
| PK15 | |||||
| Human | CaCo-2 | ||||
| Huh7 | |||||
| HepG2 | |||||
| 293 | |||||
| 293T | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siri, M.; Dastghaib, S.; Zamani, M.; Rahmani-Kukia, N.; Geraylow, K.R.; Fakher, S.; Keshvarzi, F.; Mehrbod, P.; Ahmadi, M.; Mokarram, P.; et al. Autophagy, Unfolded Protein Response, and Neuropilin-1 Cross-Talk in SARS-CoV-2 Infection: What Can Be Learned from Other Coronaviruses. Int. J. Mol. Sci. 2021, 22, 5992. https://doi.org/10.3390/ijms22115992
Siri M, Dastghaib S, Zamani M, Rahmani-Kukia N, Geraylow KR, Fakher S, Keshvarzi F, Mehrbod P, Ahmadi M, Mokarram P, et al. Autophagy, Unfolded Protein Response, and Neuropilin-1 Cross-Talk in SARS-CoV-2 Infection: What Can Be Learned from Other Coronaviruses. International Journal of Molecular Sciences. 2021; 22(11):5992. https://doi.org/10.3390/ijms22115992
Chicago/Turabian StyleSiri, Morvarid, Sanaz Dastghaib, Mozhdeh Zamani, Nasim Rahmani-Kukia, Kiarash Roustai Geraylow, Shima Fakher, Fatemeh Keshvarzi, Parvaneh Mehrbod, Mazaher Ahmadi, Pooneh Mokarram, and et al. 2021. "Autophagy, Unfolded Protein Response, and Neuropilin-1 Cross-Talk in SARS-CoV-2 Infection: What Can Be Learned from Other Coronaviruses" International Journal of Molecular Sciences 22, no. 11: 5992. https://doi.org/10.3390/ijms22115992