
Abstract
This phase 3 trial compared safety, tolerability, immunogenicity and efficacy of the self-amplifying mRNA COVID-19 vaccine, ARCT-154, with ChAdOx1-S adenovirus-vector vaccine. In four centers in Vietnam adult participants aged 18‒85 years were randomly assigned to receive two doses, 28 days apart, of either ARCT-154 (n = 1186) or ChAdOx1-S (n = 1180). Both vaccines were well tolerated with similar safety and reactogenicity profiles consisting of mainly mild-to-moderate solicited adverse events and few related serious adverse events. Higher neutralizing antibody responses persisting to one-year post-vaccination after ARCT-154 compared with ChAdOx1-S were associated with a generally higher efficacy against COVID-19. In an exploratory analysis relative vaccine efficacy of ARCT-154 vs. ChAdOx1-S against any COVID-19 from Day 36 to Day 394 was 19.8% (95% CI: 4.0–33.0). Self-amplifying mRNA vaccine offers potential immunological advantages in terms of immunogenicity and efficacy over adenovirus-vector vaccine without compromising safety.
Similar content being viewed by others


Introduction
Although the COVID-19 pandemic has abated and is no longer a WHO Public Health Emergency of International Concern (PHEIC)1 the threat of a future pandemic remains. Recurrent waves of COVID-19 cases have occurred due to emergence of new SARS-CoV-2 variants which evade the immunity of the original vaccines through accumulated mutations in the main antigenic target, the Spike glycoprotein (S protein)2. This necessitates continuation of the development of new vaccines against these variants, potentially for seasonal revaccination, applying lessons learned from rapid development of the original vaccines to improve the next generation. A variety of effective COVID-19 vaccines against the SARS-CoV-2 virus responsible for the pandemic were developed using different platforms including inactivated whole virus, mRNA, virus-vector, and recombinant protein-based vaccine that targeted the S protein of the ancestral SARS-CoV-2 virus3. Arguably, the most effective of the original vaccines were the mRNA vaccines but those vaccines have been shown to have a limited duration of effectiveness, both due to declining immunity and immune evasion4,5.
Arcturus Therapeutics, Inc (Arcturus, San Diego, CA, USA) has developed a series of self-amplifying mRNA (sa-mRNA) vaccines. After a small phase 1 demonstration of the safety and immunogenicity of a prototype sa-mRNA6 a combined Phase 1/2/3a/3b pivotal clinical study in Vietnam was initiated to demonstrate the safety and efficacy in preventing COVID-19 disease of ARCT-154, an sa-mRNA vaccine encoding the S-protein of the early Wuhan-Hu-1 virus variant, D614G7. As the study was conducted during the pandemic period and at a time when the Vietnamese national immunization campaign had been initiated with authorized COVID-19 vaccines, a switchover design was implemented on Day 92 when placebo recipients received ARCT-154 and ARCT-154 recipients received placebo to ensure all participants received COVID-19 vaccination. Of necessity this limited the duration of efficacy assessment to 92 days, 65 days post second dose. There was a concern that this time-window would be insufficient to collect a substantial number of COVID-19 cases to conclude on vaccine efficacy. Therefore, the study protocol was amended to include a Phase 3c part of the study in which immunological non-inferiority of ARCT-154 with the authorized adenovirus-vector COVID-19 vaccine developed by AstraZeneca, ChAdOx1-S, was to be assessed, together with an evaluation of the safety, reactogenicity, and efficacy of both vaccines. Here we present the immune response and reactogenicity to a primary series of two doses of ARCT-154 in comparison with ChAdOx1-S in previously unvaccinated Vietnamese adults, monitored over one year post-vaccination, together with an assessment of the relative efficacy of the two vaccines against COVID-19 at a time when the prevailing circulating strains were the Delta or Omicron variants.
Results
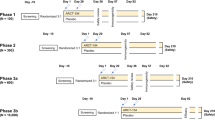
Of 3238 volunteers screened 2366 were enrolled and randomized 1:1 to the two study groups (Fig. 1). Demographics of the two groups were balanced with the same median ages (52 years) and approximately equal numbers of men and women, with similar baseline characteristics (height, weight, BMI) and equal stratification of the 18–59-year-olds into healthy and at-risk groups (Table 1).

Numbers of volunteers screened and participants enrolled in ARCT-154 and ChAdOx1-S groups at each study milestone and analysis sets are displayed. mITT modified intention-to-treat, SAS safety analysis set.
Safety
There were four deaths, all in the ChAdOx1-S group; reported during the safety follow-up period to Day 394 which were due to Non-Hodgkin’s lymphoma, gastrointestinal hemorrhage, esophageal carcinoma and a cerebrovascular accident (Table 2). The deaths were not considered to be related to study vaccination. There were 170 participants who reported at least one serious adverse event (SAE), 83 (7.0%) in 1186 ARCT-154 vaccinees and 87 (7.4%) in 1180 ChAdOx1-S vaccinees, including 8 and 9 cases of COVID-19 in each study group, respectively (Table 2). Most SAEs were not considered to be related to vaccination, but cases of angina pectoris, polyarthritis and cerebral infarction in the ARCT-154 group, and angina pectoris, chest pain and two cases of headache in the ChAdOx1-S group were considered to be related to vaccination by the investigator. Of these, two SAEs in ARCT-154 recipients (polyarthritis and cerebral infarction) and one in a ChAdOx1-S recipient (angina pectoris), resulted in their discontinuation from the study vaccinations. Medically-attended adverse events were reported by similar proportions of study participants in both vaccine groups, 461 (38.9%) and 496 (42.0%) of ARCT-154 and ChAdOx1-S groups, but most were not related to vaccination (Table 2).
After the first dose, the reactogenicities of the two vaccines were similar, with local reactions being reported by 467 (39.5%) of 1182 ARCT-154 recipients and 416 (35.4%) of the 1176 ChAdOx1-S vaccinees in the Safety set who provided data (Table 2). These local reactions mainly consisted of mild pain or tenderness at the injection site (Fig. 2), which resolved within 1–2 days. Local reactions were less frequent in both groups after the second dose, reported by 286 (24.9%) of 1150 and 183 (15.9%) of 1149 ARCT-154 and ChAdOx1-S groups (Table 2), and still consisted mainly of transient mild pain or tenderness. The frequency of severe local solicited reactions after the first or second dose was 0.1% (1/1182) in the ARCT-154 group and 0.1% (1/1176) in the ChAdOx1-S group.

The percentages of each study group reporting the solicited local reactions and systemic adverse events are displayed according to severity.
After the first dose, systemic solicited adverse events occurred in 638 (54.0%) of 1182 ARCT-154 vaccinees and 657 (55.9%) of 1176 ChAdOx1-S vaccinees. Incidence decreased to 477 (41.5%) of 1150 and 331 (28.8%) of 1149 after the second dose (Table 2). The most frequent systemic solicited adverse events were fatigue, headache, chills, and arthralgia after either dose (Fig. 2). Frequencies of severe systemic solicited AEs were similar after the first or the second doses of ARCT-154 (3.0% [35/1182]) and ChAdOx1 (3.7% [44/1176]). Solicited systemic AEs were transient, generally resolved within two days after dosing, and were similar in duration in ARCT-154 and the ChAdOx1-S groups. There were no meaningful differences in frequency or severity of adverse events observed between younger (18–59 years) and older (≥60 years) age groups, except for a lower frequency of solicited adverse reactions in older participants.
Immunogenicity
Immunogenicity assessment was performed in 917 randomly selected study participants of the immunogenicity analysis subset (IAS), 455 in the ARCT-154 group and 462 in the ChAdOx1 group, who received both study vaccinations and provided blood samples at all 5 time points (Days 1, 29, 57, 211 and 394). On Day 1, all but three participants, all in the ARCT-154 group, were negative for antibodies against the SARS-CoV-2 nucleocapsid protein (anti-N-protein), indicating no recent SARS-CoV-2 infections (Table 1). During the study period, 96 of 455 (21.1%) and 115 of 462 (24.9%) participants from the ARCT-154 and ChAdOx1-S IA subsets, respectively, reported virologically confirmed COVID-19 disease and by Day 394, only 100 of 455 (22.0%) and 76 of 462 (16.5%) participants in the ARCT-154 and ChAdOx1-S subsets, respectively, were still anti-N-protein negative (Supplementary Table 1). A further 31/100 (31%) and 23/76 (30.3%) of the individuals in the ARCT-154 and ChAdOx1-s group received non-study COVID-19 vaccines between Days 211 and 394 so were excluded from the immunogenicity analyses. This subset of N-protein-negative participants at Days 1 and 394 with no symptomatic COVID-19 constituted the SARS-CoV-2-naive cohort.
Those who seroconverted from negative to positive for anti-N-protein antibodies between Days 1 and 394—352/452 (77.9%) in ARCT-154 and 386/462 (83.5%) in ChAdOx1-S groups—with or without symptomatic COVID-19, constituted the SARS-CoV-2-exposed cohort. We randomly selected 105 participants from each vaccine group in this SARS-CoV-2-exposed cohort for immunogenicity testing of whom 10 individuals in ARCT-154 group and 17 individuals in ChAdOx1-S group had received non-study COVID-19 vaccines and were excluded from the analysis for respective time points after these non-study vaccinations.
Levels of neutralizing antibodies were either very low or unmeasurable before vaccination in both SARS-CoV-2-naive and SARS-CoV-2-exposed cohorts with baseline GMTs approximately half the LLOQ of the assay (baseline <LLOQ was imputed to half LLOQ). Four weeks after the first vaccination of SARS-CoV-2-naive participants there were marked increases in neutralizing antibodies after either vaccine, with a higher response in the ARCT-154 group—the geometric mean-fold rise (GMFR) was 7.5 (95% CI: 6.0–9.4) compared with 3.2 (2.6–3.9) after ChAdOx1-S (Fig. 3, Supplementary Table 2A). Titers increased further after the second dose, achieving GMFR of 29.9 (24.6–36.2) in the ARCT-154 group vs. 11.0 (8.7–13.9) in the ChAdOx1-S group. The ratios of GMTs between ARCT-154 and ChAdOx1-S groups were 2.34 (95% CI: 1.71–3.21) on Day 29 and 2.70 (2.00–3.64) on Day 57. On Day 211, 6 months after the second vaccination, the GMT plateaued in these subsets with no natural exposure to SARS-CoV-2—126 (95% CI: 86.2–183) after ARCT-154 and 77.5 (48.3–124) after ChAdOx1-S (Fig. 3). In SARS-CoV-2-naive participants the GMT increased to 253 (153–416) by Day 394, one year after vaccination, in the ARCT-154 group but was unchanged at 68.4 (40.8–115) in the ChAdOx1-S group, giving an ARCT-154 vs. ChAdOx1-S GMT ratio of 3.70 (1.80–7.59) (Fig. 3). Non-inferiority criteria were met for ARCT-154 compared with ChAdOx1-S at all post-vaccination time points.

Geometric mean titers of neutralizing antibody responses against Wuhan-Hu1 strain measured by CPE-based microneutralization assay (1/dil) after doses 1 (Day 1) and 2 (Day 29) of ARCT-154 or ChAdOx1-S, in baseline anti-N-protein naive subjects, according to N-protein seropositivity status on Day 394. Chart shows geometric mean titers (95% CI) with n values for each time point, and table shows the GMT ratio between ARCT-154 and ChAdOx1-S groups and the geometric mean-fold rises at each time point (with 95% CI).
In the SARS-CoV-2-exposed cohort the immune response to the first vaccination was also higher in the ARCT-154 group than in the ChAdOx1 group with GMFR of 7.9 (95% CI: 6.2–10.0) and 5.1 (4.0–6.4), respectively, and the inter-group GMT ratio of 1.56 (95% CI: 1.13–2.17) (Fig. 3, Supplementary Table 2B). The GMTs increased further after the second dose with GMFRs of 34.2 and 15.4 in ARCT-154 and ChAdOx1-S groups on Day 57, a GMT ratio of 2.22 (95% CI: 1.61–3.07), and continued to increase in both groups, presumably due to natural exposure to SARS-CoV-2. On Days 211 and 394 the inter-group GMT ratios were 1.31 (95% CI: 0.80–2.14) and 1.87 (1.30–2.71).
Age of the participants had no impact on the immune responses in SARS-CoV-2-naive or exposed participants (Supplementary Fig. 1), with responses similar in those aged 18–59 years, or those aged 60 years or more. The effect of apparent SARS-CoV-2 infection was still observed, with a continuing increase in GMTs up to Day 394, while in SARS-CoV-2-naive participants the GMTs plateaued at Days 211 and 394.
We assessed cross-neutralizing antibodies against SARS-CoV-2 Delta, Omicron BA.2, XBB.1.5.6 and BA.2.86 subvariants in a subset of participants (n = 31) from the ARCT-154 group who met the following criteria: were anti-N-protein negative on Day 1, did not have a virologically confirmed COVID-19 infection during the study, and had not received other COVID-19 vaccines. On Day 57, GMTs of neutralizing antibodies against the Wuhan strain were 485 (95% CI: 347–678), 34 (22–52) against Delta strain, and 16 (12–21) against Omicron BA.2 (Supplementary Table 3). Titers of neutralizing antibodies against more recent Omicron subvariants XBB.1.5.6 and BA.2.86 were close to the assay LLOQ.
The majority of these participants, 81% (25 of 31), seroconverted for anti-N-protein antibodies at Day 394, indicating asymptomatic infection or exposure to SARS-CoV-2 (Supplementary Table 3). Such natural infection elicited a robust booster response in neutralizing antibodies against all tested variants, most prominently for Omicron BA.2 with GMT increasing from 15 (95% CI: 12–19) to 756 (483–1182), then Delta from 30 (20–45) to 399 (223–715), and Wuhan-Hu-1 from 453 (312–656) to 2982 (2028–4384). Smaller increases in antibody titers were observed for Omicron XBB.1.5.6 subvariant from 11 (10–13) to 81 (46–142) and for Omicron BA.2.86 from 10 (10–11) to 24 (17–35).
Efficacy assessment
Overall, 48 confirmed cases of COVID-19 were reported between Days 36 and 92, but this was followed by a significant surge in cases up to the end of the study by when there was a total of 480 cases. Of these, 450 cases occurred between Days 36 and 211, with a further 30 cases then occurring up to Day 394. No case of severe COVID-19 was reported by the investigators or identified during the blinded case reviews, but six participants with COVID-19 infections were hospitalized for isolation purposes. The most frequent clinical symptoms in participants with COVID-19 were cough (58.3%), fever/chills (50.4%), sore throat (40.2%), headache (36.0%) and fatigue (34.6%). No sequencing was performed in this part of the study to identify variants of SARS-CoV-2 but epidemiological data are available for the study period showing the main circulating SARS-CoV-2 strains were Omicron sub-lineages BA.2 and BA.5 (Supplementary Fig. 2) and occurrence of COVID-19 cases and associated deaths (Supplementary Fig. 3).
In mITT participants, 20 and 28 virologically confirmed COVID-19 cases were reported in ARCT-154 and ChAdOx1-S groups, respectively, from Day 36 to Day 92, the end of per-protocol efficacy surveillance in the previous Phase 1/2/3a/3b part of the study (Table 3, Fig. 4A). This equates to a relative vaccine efficacy (rVE) for ARCT-154 of 30.7% (95% CI: −23.0–61.0) over a period when predominant SARS-CoV-2 strains were Delta (B.1.617.2) and Omicron BA.1 variants (Supplementary Fig. 2). As monitoring in this phase 3c study continued, the confirmed cases increased to 205 and 245 in ARCT-154 and ChAdOx1-S groups, respectively, during six months post-vaccination (until Day 211) due to a new surge of COVID-19 cases caused by the Omicron BA.2 variant (Supplementary Fig. 2). During this period the positive difference in rVE in favor of ARCT-154 over ChAdOx1-S of 19.3% (95% CI: 2.8–32.9) persisted (Table 3, Fig. 4B). For the full one year of monitoring from Day 36 until Day 394, the totals of 218 and 262 cases reported in ARCT-154 and ChAdOx1-S groups gave a rVE of 19.8% (95% CI: 4.0–33.0) (Table 3). When analyzed according to age group there is a trend for the rVE of ARCT-154 vs. ChAdOx1-S to increase with age (Table 3), despite no age-dependent difference being observed in the immune response.

Relative vaccine efficacy (rVE) of ARCT-154 vs. ChAdOx1-S vaccine against any severity of COVID-19 from Day 36 (one week after the second dose) up to Day 92 (A) and from Day 36 up to Day 211 (B).
Discussion
The COVID-19 pandemic resulted in the rapid development of a multitude of different vaccines of different types using different manufacturing platforms3. Some of the most successful were based on mRNA coding for the Spike glycoprotein (S-protein) of the SARS-CoV-2 virus. The mRNA is delivered using lipid nanoparticles formulations to protect the integrity of mRNA. An alternative vaccine platform used is the fusion of the S-protein mRNA in a viral-vector vaccine which transports the mRNA into the cell, a strategy which has been applied to develop vaccines against Ebola, RSV, Zika virus as well as SARS-CoV-2. Following a different strategy Arcturus developed a self-amplifying mRNA (sa-mRNA) system in which an RNA-dependent RNA polymerase replicates the RNA coding for the S-protein antigen. Theoretical advantages of sa-mRNA vaccines include use of much smaller quantities of mRNA to elicit equivalent immune responses compared with the conventional mRNA vaccines, as well as larger payloads, extended protein expression and longer durability of immune response8,9,10.
The present study was performed as part of a larger phase 1/2/3 clinical assessment of ARCT-154, the Arcturus sa-mRNA vaccine candidate, encoding the Wuhan-Hu-1 SARS-Cov-2 D614G virus. This combined study included a placebo-controlled assessment of the efficacy of two doses of ARCT-154, administered on Days 1 and 29, against COVID-19 disease from Day 36, 7 days after the second dose, until Day 927. Over this protocol-defined surveillance period ARCT-154 had a vaccine efficacy (VE) of 56.6% (95% CI: 48.7–63.3) against any COVID-19 disease despite the dominant circulating strain at the time of the assessment being the Delta variant; VE against severe COVID-19 was 95.3% (80.5–98.9)7. The current phase 3c adjunct to that study was intended to compare the safety and demonstrate non-inferiority of the immune response of ARCT-154 with the licensed adenovirus-vector vaccine, ChAdOx1-S, together with an exploratory assessment of efficacy against COVID-19. However, between 3 and 6 months after initiation of this Phase 3c study there was a surge in COVID-19 disease incidence in Vietnam caused by the Omicron BA.2 variant (Supplementary Fig. 2) resulting in a significant increase in the number of symptomatic and asymptomatic COVID-19 disease in both study groups, affecting more than 78% of the study population (Supplementary Table 1). With this major surge in COVID-19 infection changing the background epidemiology of SARS-CoV-2 we did not pursue the non-inferiority assessment of immunogenicity due to such high levels of natural exposure.
Serological testing for N-protein, as already implemented on Day 1, was also performed on Day 394 for all participants in the immunogenicity subset who had or had not displayed any evidence of symptomatic COVID-19 during the study to determine who had asymptomatic infection. This found over half of these participants had evidence of seroconversion to N-protein without symptoms of SARS-CoV-2 (Supplementary Table 1).
We therefore assessed the immune response to vaccination both in those who apparently did not have any natural exposure to SARS-CoV-2 and so remained seronegative for N-protein throughout, and in those with evidence of asymptomatic SARS-CoV-2 infection, which would lead to hybrid immunity11. We were able to show that the neutralizing antibody responses following a first or second primary dose of ARCT-154 were non-inferior, indeed greater than those observed after ChAdOx1-S. Further, we assessed the persistence of these responses out to one year after vaccination and found better persistence after ARCT-154 than ChAdOx1-S, in both SARS-CoV-2-naive and SARS-CoV-2-exposed cohorts. Similar results have been observed in Japan, where a booster dose of ARCT-154 was directly compared with a booster dose of the mRNA vaccine, BNT162b2, in mRNA-primed adults and found to elicit a higher immune response against both Wuhan-Hu-1 and Omicron BA.4/5 variant12. The neutralizing antibody response after ARCT-154 was not only higher in magnitude than after BNT162b2 but was also more persistent up to six months after the booster dose13. Importantly, the asymptomatic infection (during the Delta and Omicron BA.2 surges) in previously vaccinated individuals induced a broadly neutralizing booster response against historical strains (Wuhan, Delta, Omicron BA.2) but also elicited some cross-protective antibodies against future variants (XBB.1.5.6 and BA.2.86). In the present study, the small variations in GMTs in SARS-CoV-2-naive participants at Days 211 and 394 may have been due to some natural exposure to SARS-CoV-2 variants, which did not lead to full symptomatic or asymptomatic infections detectable by N-protein serology which assay is known to have a relatively low sensitivity14.
When assessed over the period from Day 36 to Day 92, as in the efficacy analysis in the phase 1/2/3a/3b part of the study there was a relatively small number of verified COVID-19 cases (Fig. 4A), 20 and 28 in ARCT-154 and ChAdOx1-S groups, respectively giving an rVE of 30.7% (95% CI: −23.0–61.0) in favor of ARCT-154. Although we did not assess the strains responsible for COVID-19 cases in our study population, the national surveillance in Vietnam showed that during this Day 36 to 92 period the predominant circulating SARS-CoV-2 strain in Vietnam were the Delta (B.1.617.2) and Omicron BA.1 variants (Supplementary Fig. 2). The incidence of SARS-CoV-2 infections then increased substantially from around Day 100, following the national emergence of the Omicron BA.2 sub-lineage which led to a marked spike in occurrence of cases (Supplementary Fig. 3). This was followed by emergence of the Omicron BA.5 sub-lineage (Supplementary Fig. 2) which in itself did not increase the frequency of cases. Despite the marked increase in cumulative incidence there was still an rVE of 19.3% (95% CI: 2.85–32.9) in favor of ARCT-154 over ChAdOx1-S when assessed from Day 36 to Day 211, the difference persisting as an rVE of 19.8% (4.0–33.0) when assessed over the period from Day 36 to Day 394.
Lower protective efficacy against Delta and Omicron variants has been observed with all COVID-19 vaccines based on the ancestral Wuhan-Hu-1 strain15. In a test-negative case-control study in a Canadian population vaccinated with ChAdOx1-S or mRNA vaccines, the estimated vaccine effectiveness against symptomatic Omicron infection 7–59 days after a second dose was 36% (95% CI: 24–45) which decreased to 1% (95% CI: −8–10) after 180 days or longer. When measured 7 or more days after a third (booster) dose, VE increased to 61% (95% CI: 56–65). Vaccine effectiveness against severe COVID-19 outcomes remained high after a two-dose primary series and a booster dose15.
Safety and reactogenicity of ARCT-154 appeared to be at least comparable with ChAdOx1-S although the size of our study precludes us from being able to detect any rare adverse event associated with vaccination. As with mRNA vaccines, ChAdOx1-S has been shown to be associated with rare side-effects including myocarditis, acute disseminated encephalomyelitis (ADEM) and Guillain-Barré syndrome (GBS) which are only detectable in populations in which millions of doses have been administered16. Only widespread use of ARCT-154 will reveal whether there are any similar safety concerns, but it should be noted that such events usually resolve without sequelae and are more frequent following SARS-CoV-2 infection and COVID-19 disease rather than vaccination17.
Our study was limited in that it was not a placebo-controlled trial, as it was an adjunct to the larger placebo-controlled phase 1/2/3a/3b trial that demonstrated the efficacy of the vaccine. With no placebo arm an assessment of absolute VE of both vaccines was not possible, but our observations with ARCT-154 and ChAdOx1-S were similar, with no severe COVID-19 cases observed, and no hospitalizations for treatment of COVID-19 symptoms during Delta and Omicron circulation. The selection of participants with asymptomatic SARS-CoV-2 infection based on seroconversion for anti-N-protein antibodies has some limitations as the anti-N-protein antibody response can be transient14, and with limited assay sensitivity not all participants with asymptomatic infection may have been detected with this test. However, in the current study, this selection bias was not fully eliminated but addressed by the presence of an active control group. In general, comparisons of ARCT-154 with the ChAdOx1-S vaccine tended to be favorable for ARCT-154 although as indicated above, no clinical trial is anticipated to be able to detect extremely rare adverse events associated with some COVID-19 vaccines. The occurrence of a surge of SARS-CoV-2 infections due to Omicron variants during the study did not allow us to pursue our non-inferiority comparison of ARCT-154 vs. ChAdOx1-S, but a separate study in Japan has shown the superior immunogenicity of ARCT-154 compared with the BNT162b2 mRNA vaccine both in terms of magnitude and persistence of immune response12,13. We only assessed the neutralizing antibody responses and did not include any analyses of the other possible components of the immune response, e.g. cell-medicated immunity. Such factors are being investigated in other studies of ARCT-154.
We have shown that ARCT-154 is as well tolerated and at least as immunogenic and effective as the licensed vaccine, ChAdOx1-S, in protecting against severe COVID-19 disease in a period when the circulating SARS-CoV-2 viruses were Delta and sub-lineages of the Omicron variants. These data, together with the demonstrated superior responses to ARCT-154 compared with licensed vaccines in other studies suggest that sa-mRNA could be a valuable addition to the existing COVID-19 vaccines to maintain protection against future COVID-19 pandemics due to emerging variants.
Methods
This phase 3c randomized, observer-blind, controlled study was performed as part of a larger phase 1/2/3 study investigating primary series and booster vaccination with ARCT-154 vaccine7. That study is registered with ClinicalTrials.gov (identifier: NCT05012943). Recruitment for this phase 3c part was done in four clinical centers in Vietnam from 1 to 8 November 2021; database lock for the final phase 3c analysis was 15 August 2023. The protocol was approved by the ethics committee of each study center and the Vietnam National Ethical Committee in Biomedical Research and Ministry of Health, and the overall study was conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki and International Council for Harmonisation (ICH E6R2) guidelines, with applicable local regulatory and bioethics requirements. The primary objectives of this phase 3c study were to assess the safety and reactogenicity of ARCT-154 and ChAdOx1-S and to demonstrate non-inferiority of the immune response to ARCT-154 vs. ChAdOx1-S assessed as surrogate neutralizing antibodies at Day 57, four weeks after a second vaccination; secondary objectives included superiority of the immunogenicity of ARCT-154 vs. ChAdOx1-S and an exploratory assessment of the relative vaccine efficacy against virologically confirmed COVID-19.
Participants
Eligible participants were healthy males or females aged 18 years or older who had not previously received any COVID-19 vaccination and were willing and able to consent to all study procedures. Participants were stratified into two age groups, 18–59 years of age and ≥60 years of age, with 18–59-year-olds selected to equally represent those considered healthy and those at high risk of severe COVID-19 due to underlying co-morbidities using the US Centers for Disease Control and Prevention (CDC) definitions current at the time of the study18. Exclusion criteria included any evidence of an acute infection at the time of enrollment, any previous COVID-19 infection (including a positive result of RT-PCR), close contact with a person known to be infected with SARS-CoV-2, or any known history of anaphylactic reactions to vaccines. Women were not to be pregnant or breastfeeding and were required to use an approved birth control method for 2 months after vaccination. Detailed exclusion criteria are shown in Supplementary information file.
Procedures and safety assessments
At enrollment on Day 1, after having the study explained to them and signing an informed consent form, participants were randomized 1:1 via Interactive Voice Response System (IVRS) to two study groups to receive ARCT-154 or ChAdOx1. Prior to randomization, participants were stratified by the following factors: age <60 at high risk of severe COVID-19, age < 60 not at high risk of severe COVID-19, age ≥60 years of age (considered at risk of severe COVID-19 by default). All study participants provided a pre-vaccination blood sample before receiving a dose of their assigned study vaccine. The procedure was repeated on Day 29 for the administration of the second vaccination. All subsequent procedures were done by study personnel blinded to study group. Participants were monitored for 30 minutes for any immediate reactions after each vaccination and then supplied with diary cards to record the occurrence and severity of solicited local reactions and systemic adverse events (see Supplementary Tables 4 and 5) for 7 days after each vaccination and to record any unsolicited adverse events within 28 days after each study vaccination. Data about the occurrence, severity, and relatedness to vaccination of any serious adverse events (SAE) and medically-attended adverse events (MAAE) were collected during the entire study period (from Day 1 to Day 394). Sera were obtained from all participants at baseline to measure the presence of antibodies against the SARS-CoV-2 nucleocapsid protein (N-protein) to assess whether a recent natural infection had occurred. Additional sera were collected from designated participants in the immunogenicity subset on Days 1, 29, 57, 211 and 394 to assess the initial immune response as well as antibody persistence.
Vaccines
The study vaccine was ARCT-154 (Batch 159568 A, Arcturus Therapeutics Inc. San Diego, CA, USA) consisting of a replicon based upon Venezuela equine encephalitis RNA virus in which the virus structural proteins segment is replaced with RNA coding for the full-length spike glycoprotein of the SARS-CoV-2 D614G variant. ARCT-154 was supplied in vials containing 100 μg active ingredient encapsulated in lipid nanoparticles stored at −20 °C or lower. Immediately before use, the vial contents were diluted in 10 mL sterile saline, so each 0.5 mL dose contained 5 μg of mRNA. The comparator vaccine, ChAdOx1-S (Batches A1093 or ACA3094, AstraZeneca) is a chimpanzee adenovirus encoding the SARS-CoV-2 spike glycoprotein and was prepared according to the manufacturer’s instructions. Each 0.5 mL dose contains not less than 2.5×108 infectious units for intramuscular injection. Both vaccines were prepared in identical syringes and administered by unblinded study personnel as intramuscular injections in the deltoid muscle so the participant and other study personnel remained blinded.
Immunogenicity
All baseline blood samples were tested for antibodies against the SARS-CoV-2 nucleocapsid protein (N-protein), a positive result being considered as indicative of previous infection as the N-protein was not included in any vaccine administered in Vietnam during the study period. Sera from the immunogenicity subset were sent to a central laboratory (VisMederi, Siena, Italy) to measure neutralizing antibody titers using a validated virus cytopathic effect (CPE)-based micro neutralization assay with results expressed in titers (reciprocal of the lowest dilution causing 50% neutralization). The primary analysis was done with the parental Wuhan-Hu-1 strain, but additional characterizations were also done for Delta, Omicron BA.2, XBB.1.5.6 and BA.2.86 subvariants in a subset of participants from the ARCT-154 group. Group titers were expressed as geometric mean titers (GMT), seroconversion rates (SCR; seroconversion being a four-fold increase in titer over baseline), and geometric mean fold rises (GMFR) from Day 1, with samples below the lower limit of quantitation (LLOQ, lower than 1:20 dilution) being assigned a value of 10 (half the LLOQ). In addition, all Day 394 samples from immunogenicity subset were tested for antibodies against N-protein, to identify natural SARS-CoV-2 infection during the study period.
Efficacy assessment
From three days after the first vaccination participants with potential symptoms and clinical signs of COVID-19 (fever, chills, cough, shortness of breath or difficulty breathing, fatigue, muscle or body aches, headache, new loss of taste or smell, sore throat, congestion or runny nose, nausea, vomiting or diarrhea) were evaluated for the presence of COVID-19. Where possible, participants visited their respective study centers where nasal swabs were taken for RT-PCR testing and clinical symptoms/signs and associated medications were recorded. A protocol-defined COVID-19 case had to have virological confirmation (by RT-PCR or, in a few cases by Rapid antigen test) of SARS-CoV-2 and at least one of the symptoms or clinical findings listed above. Case definitions to evaluate the severity of COVID-19 were based on US FDA recommendations; severe COVID-19 included any of the following: clinical signs at rest indicative of severe systemic illness, acute respiratory failure, evidence of shock, cardiac, renal, hepatic, or neurologic dysfunction; admission to an intensive care unit, or death (Supplementary Table 6). An internal, blinded review of all suspected COVID-19 cases was performed and concluded on whether the case met the protocol-defined COVID-19 case criteria and whether the case met the criteria of severe COVID-19 according to the US FDA. Only virologically confirmed, protocol-defined and independently evaluated cases were included in the vaccine efficacy (VE) analysis.
Statistics
Safety assessments were analyzed in the safety analysis set (SAS), comprising all participants who received at least one dose of assigned vaccine, and reactogenicity analysis sets (RAS), consisting of all SAS participants who completed diary cards. The primary safety endpoints were any solicited local or systemic AEs starting within 7 days after each vaccination by severity grade and any unsolicited AE starting within 28 days after each vaccination, summarized by severity and relationship to study vaccine as well as any medically-attended adverse event (MAAE), serious adverse event (SAE), or AE leading to withdrawal or discontinuation up to the end of the study. There was no primary hypothesis for safety testing, comparisons being made with estimates and 95% confidence intervals (CI). With a sample size of approximately 2400 participants and approximately 1200 participants randomized to receive ARCT-154 or ChAdOx1-S for the safety analysis, based upon the following formula, if the incidence rate of an AE was 0.1%, the probability to detect one event in 1200 vaccinated participants would be 69.9% and if the incidence rate was 1.0% the probability would be more than 99%: p = 1−(1−R)N for an incidence rate of R and sample size of N.
The population for the immunogenicity analysis subset (IAS) consisted of the first 1500 participants enrolled. The primary immunogenicity endpoint was the neutralizing antibody response evaluated on Day 57 as the geometric mean titer (GMT) for each study group and the geometric mean titer ratio (GMR) of ARCT-154 vs. ChAdOx1-S groups. Assuming a GMR ≥ 0.9 and a coefficient of variation of 4.6, a sample size of 800 participants would provide greater than 90% power to exclude a noninferiority boundary of 0.67 based on the use of a one-sided test at the alpha = 0.025 level of significance, accounting for a dropout rate of approximately 20–25% due to the national COVID-19 vaccination campaign in Vietnam. Immunogenicity analyses were performed on both SARS-CoV-2-naive and SARS-CoV-2-exposed cohorts based on history of exposure to SARS-CoV-2 evidenced by N-protein serology and medical history of confirmed symptomatic COVID-19 during the study.
The exploratory efficacy objective, relative Vaccine Efficacy (rVE), was calculated in the modified Intention to Treat (mITT) set defined as all participants who received both doses of the assigned study vaccine and had no clinical or serologic evidence of SARS-CoV-2 infection up to Day 36, 7 days after the second study injection. The main endpoint was the first occurrence of confirmed, protocol-defined COVID-19 with onset after Day 36 and rVE was defined as % reduction in the hazard rate of the COVID-19 events (ARCT-154 vs ChAdOx1-S) = 1 − ℎ (ARCT-154)/ℎ (ChAdOx1-S) = 1 − HR, where h(ARCT-154) and h(ChAdOx1-S) are hazard rates of COVID-19 events among those in ARCT-154 and ChAdOx1-S groups, respectively. A Cox proportional hazard model was used to assess the magnitude of the study group difference (i.e., Hazard Ratio) between ARCT-154 and ChAdOx1-S at a 1-sided 0.025 significance level. Factors used as covariates in the Cox proportional hazard regression included: Risk group: ≥18 to <60 years and “healthy”, ≥18 and <60 years and “at risk of severe COVID-19” and ≥ 60 years and study site region.
Data availability
After the final study report is prepared, the data generated in this study will be made available to suitably qualified scientific researchers who make a request to the senior investigator or study sponsor with a appropriate protocol for a valid research project.
References
Lenharo M. WHO declares end to COVID-19’s emergency phase. Nature (published online May 5) https://doi.org/10.1038/d41586-023-01559-z (2023).
Mengist, H. M. et al. Implications on immune evasion and vaccine-induced immunity. Semin. Immunol. 55, 101533 (2021).
Nagy, A. & Alhatlani, B. An overview of current COVID-19 vaccine platforms. Comp. Struct. Biotechnol. J. 19, 2508–2517 (2021).
Menegale, F. et al. Evaluation of waning of SARS-CoV-2 vaccine-induced immunity: a systematic review and meta-analysis. JAMA Netw. Open 6, e2310650 (2023).
Andrejko, K. L. et al. Waning of 2-dose BNT162b2 and mRNA-1273 vaccine effectiveness against symptomatic SARS-CoV-2 infection accounting for depletion-of-susceptibles bias. Am. J. Epidemiol. 192, 895–907 (2023).
Low, J. G. et al. A phase I/II randomized, double-blinded, placebo-controlled trial of a self-amplifying Covid-19 mRNA vaccine. npj Vaccines 7, 161 (2022).
Hồ, N. T. et al. Safety and immunogenicity and efficacy of the self-amplifying mRNA ARCT-154 COVID-19 vaccine; pooled phase 1, 2, 3a and 3b randomized, controlled trials. Nat. Commun. 15, 4081 (2024).
Bloom, K., van den Berg, F. & Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 28, 117–129 (2021).
Lundstrom, K. The potential of self-amplifying RNA vaccines for infectious diseases and COVID-19. Vaccin. Res. 7, 25–37 (2020).
Vogel, A. B. et al. Self-amplifying RNA vaccines give equivalent protection against influenza to mRNA vaccines but at much lower doses. Mol. Ther. 26, 446–455 (2018).
Crotty, S. Hybrid immunity. Science 372, 1392–1393 (2021).
Oda, Y. et al. Immunogenicity and safety of a booster dose of a self-amplifying RNA COVID-19 vaccine (ARCT-154) versus BNT162b2 mRNA COVID-19 vaccine: a double-blind, multicentre, randomised, controlled, phase 3, non-inferiority trial. Lancet Infect. Dis. 24, 351–360 (2024).
Oda, Y. et al. Persistence of immune responses of a self-amplifying RNA COVID-19 vaccine (ARCT-154) versus BNT162b2. Lancet Infect. Dis. 24, 341–343 (2024).
Krutikov, M. et al. Prevalence and duration of detectable SARS-CoV-2 nucleocapsid antibodies in staff and residents of long-term care facilities over the first year of the pandemic (VIVALDI study): prospective cohort study in England. Lancet Healthy Longev. 3, e13–e21 (2022).
Buchan, S. A. et al. Estimated effectiveness of COVID-19 vaccines against Omicron or delta symptomatic infection and severe outcomes. JAMA Netw. Open 5, e2232760 (2022).
Faksova K. et al. COVID-19 vaccines and adverse events of special interest: a multinational global vaccine data network (GVDN) cohort study of 99 million vaccinated individuals. Vaccine 42, 220–2211 (2024).
Patone, M. et al. Risks of myocarditis, pericarditis, and cardiac arrhythmias associated with COVID-19 vaccination or SARS-CoV-2 infection. Nat. Med. 28, 410–422 (2022).
CDC. Underlying medical conditions associated with high risk for severe COVID-19: information for healthcare providers/summary of conditions with evidence. https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-care/underlyingconditions.html (2023).
Acknowledgements
The authors are grateful to the study staff at Hanoi Medical University and Pasteur Institute Hochiminh City for assisting with study conduct, and at the VisMederi Laboratory (Siena, Italy) and the Pasteur Institute Nha Trang for immunogenicity assays; VietStar Biomedical Research for participating in study conduct and management; Medprove and BIOPHICS for data management. We thank Keith Veitch (keithveitch communications, Amsterdam, The Netherlands) for editorial assistance in the preparation of the manuscript. This study was co-funded by Vinbiocare Biotechnology Joint Stock Company (Hanoi, Vietnam) and Arcturus Therapeutics Inc. (CA, USA).
Author information
Authors and Affiliations
Contributions
N.T.H., X.-H.N., L.T.L.T., V.T.N., and S.G.H. participated in the design, protocol development, and conduct of the study. N.T.H., H.T.P., X.L., R.B., and I.S. participated in verifying the underlying data reported in the manuscript. N.T.H., C.V., R.B., and I.S. oversaw and participated in the data analysis plan, data analysis, and manuscript preparation. V.T.N., X.-H.N. had overall management of the study. X.-H.N., and R.S. oversaw laboratory testing and analyses. L.T.L.T., A.T.V.L., A.N.N., and D.B. oversaw study operations. V.T.T., A.T.V.P., T.Vu.N., and Q.C.L. oversaw the study conducted at the sites.
Corresponding author
Ethics declarations
Competing interests
S.G.H., R.B., X.L., C.V., R.S., D.B.., and I.S. are or were all full-time employees of the vaccine manufacturer and study sponsor, Arcturus Therapeutics, Inc. T.T.L.L. and N.T.V. are employees of the vaccine licensee, Vietnam Biocare Biotechnology Jointstock Company. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ho, N.T., Hughes, S.G., Sekulovich, R. et al. A randomized trial comparing safety, immunogenicity and efficacy of self-amplifying mRNA and adenovirus-vector COVID-19 vaccines. npj Vaccines 9, 233 (2024). https://doi.org/10.1038/s41541-024-01017-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-024-01017-5
