
Abstract
Variants of SARS-CoV-2 have continued to emerge across the world and cause hundreds of deaths each week. Due to the limited efficacy of vaccines against SARS-CoV-2 and resistance to current therapies, additional anti-viral therapeutics with pan-coronavirus activity are of high interest. Here, we screen 2.8 billion compounds from a DNA-encoded chemical library and identify small molecules that are non-covalent inhibitors targeting the conserved 3CL protease of SARS-CoV-2 and other coronaviruses. We perform structure-based optimization, leading to the creation of a series of potent, non-covalent SARS-CoV-2 3CL protease inhibitors, for coronavirus infections. To characterize their binding mechanism to the 3CL protease, we determine 16 co-crystal structures and find that optimized inhibitors specifically interact with both protomers of the native homodimer of 3CL protease. Since 3CL protease is catalytically competent only in the dimeric state, these data provide insight into the design of drug-like inhibitors targeting the native homodimer state. With a binding mode different from the covalent 3CL inhibitor nirmatrelvir, the protease inhibitor in the COVID drug Paxlovid, these compounds may overcome resistance reported for nirmatrelvir and complement its clinical utility.
Similar content being viewed by others
Introduction
SARS-CoV-2 is the viral pathogen responsible for the global COVID-19 pandemic, which has caused millions of infections and deaths worldwide. Although the weekly death toll is now lower than it was in March 2020, more than a hundred deaths are still reported each week1,2. The ongoing impact of COVID is also reflected in thousands of patients who are being treated for the disease in intensive care units and millions of others who are dealing with symptoms of long COVID. Meanwhile, variants of SARS-CoV-2 and other coronaviruses continue to be a major health threat, especially for those age 65 and older. Considering the limited effectiveness of current vaccines against the evolving variants of SARS-CoV-2, and the concern that additional coronavirus pandemics may emerge in the future, additional direct-acting anti-viral therapeutics are still in demand3.
We previously optimized the structure of the feline coronavirus inhibitor GC376 and developed a small molecule SARS-CoV-2 3CL protease inhibitor termed coronastat (NK01-63) as a candidate for COVID-19 treatment4. This is based on the essential role of 3CL protease (Mpro) in the maturation and replication of coronaviruses such as SARS-CoV-2, which makes it an attractive target for COVID-19 therapy5,6. However, further examination of coronastat in animal studies revealed that it had activity against human proteases and intrinsic reactivity as a covalent inhibitor, which limited its biodistribution, stability, and selectivity. Nirmatrelvir, the primary active ingredient of the COVID-19 drug Paxlovid, is a SARS-CoV-2 3CL protease inhibitor7. Although nirmatrelvir is clinically useful for COVID patients at risk of severe disease, the drug has to be co-administered with ritonavir in Paxlovid, to inhibit metabolic degradation of nirmatrelvir8. For covalent inhibitors such as nirmatrelvir, concern for target selectivity and pharmacokinetic (PK) profiles derive from the intrinsic reactivity of covalent warheads9. Furthermore, multiple pathways to resistance to nirmatrelvir have been reported in vitro10 and several 3CL protease mutations (L50V, E166V/A, and T169S) that confer resistance to nirmatrelvir have been observed in the clinic11,12. These concerns highlight a need for developing next generation 3CL protease inhibitors.
DNA-encoded chemical library (DEL) technology represents a powerful tool to discover small molecules capable of reversibly binding to a target protein. A DEL contains compounds with DNA tags. By applying combinatorial chemistry, building of a DEL starts with an oligonucleotide that contains a linker to a chemical building block moiety, and proceeds through iterative cycles of DNA barcode elongation and conjugation of chemical building blocks onto the linker moiety. For instance, if there are 1,000 different building blocks in each iterative cycle, three iterations will theoretically generate one billion different combinations of tagged compounds. Therefore, DEL technology enables low-cost mass creation and screening of chemical libraries of billions of compounds, increasing the chance of discovering hits. Conjugation with DNA tags significantly accelerates the screening process, as DEL screening can be performed in one large pool. Screening of the entire DEL is typically done in a single tube, in which each small molecule is represented in minute quantities. After a minimal amount of target protein is added to the tube, compounds with high affinity bind to the target protein before its immobilization onto beads, while those that don’t bind are washed away. Strong reversible binders can then be eluted and decoded by reading DNA tags, similar to scanning barcodes.
In this work, we undertake a de novo approach to screen a DNA-encoded chemical library for non-covalent inhibitors specifically targeting the coronavirus 3CL protease. Structure-based optimization of the top screening hit leads to the discovery of compounds that function as potent and selective 3CL protease inhibitors and exhibit antiviral activity on HCoV-229E, HCoV-OC43, SARS-CoV, MERS-CoV, and SARS-CoV-2.
Results
Identification of SARS-CoV-2 3CL protease inhibitors from DNA-encoded chemical library screening
Aiming to screen 2.8 billion compounds in a DNA-encoded chemical library (DELopen by WuXi AppTec) for SARS-CoV-2 3CL protease inhibitors, we captured His-tagged 3CL protease on Ni-NTA magnetic beads for binding affinity screening (Supplementary Fig. 1). After incubation with the DELopen library, 3CL protease beads or control empty beads were magnetically pulled down and washed before being heated to 95 °C to release bound library molecules.
In the first screen, four compounds (DEL1 through DEL4) emerged as top screening hits with consistently high enrichment scores in three replicates for binding to 3CL protease (Fig. 1a). We designed a counterscreen to distinguish between compounds binding at the same binding site as known inhibitors or different binding sites. In this second screen, four copies of the DELopen library were separately screened against beads capturing 3CL alone, the 3CL-GC376 complex, the 3CL-Compound 4 complex, or no protein as a negative control. GC376 and Compound 4 are known 3CL inhibitors described in previous studies4,13,14. This counterscreen enables identification of protein binders with the same binding site as the known inhibitors, because they are expected to bind less efficiently to the protein-inhibitor complex than to the protein alone. Meanwhile, small molecules with binding sites different from the known inhibitors are expected to bind to the protein-inhibitor complex and to the protein alone in an indistinguishable manner.

A Enrichment scores of top screening hits (DEL1 through DEL4) on three replicates of DELopen for binding to 3CL protease. B Enrichment scores of top screening hits (DEL5 through DEL8) in a counterscreen against beads capturing 3CL alone, the 3CL-GC376 complex, and the 3CL-Compound 4 complex, to distinguish between compounds that share binding sites with known inhibitors or target different binding sites. C Structures of top screening hits DEL1 through DEL8. For close analogs of DEL7, structure differences from DEL7 are shown in red color.
Four compounds (DEL5 through DEL8) emerged as top screening hits with enrichment for 3CL alone compared to the 3CL-GC376 complex, the 3CL-Compound 4 complex, and the negative control (Fig. 1b, c). Since both GC376 and Compound 4 are known to bind to the active site of the 3CL protease, DEL5 through DEL8 are likely to interact with the active site as well. No compound was found to be enriched for the protein-inhibitor complex or the protein alone, suggesting the known active site might be the most suitable binding site for small molecules on the surface of 3CL protease, and that additional allosteric binding sites are not likely to exist or to be easy to target.
Our further evaluation of these screening hits started from measuring their enzymatic inhibition potency using purified 3CL protease in vitro. We found that DEL2, DEL7, DEL1, DEL3, and DEL8 inhibited the activity of the 3CL protease on a specific fluorogenic substrate, with IC50 values of 0.3 µM, 0.3 µM, 4 µM, 4 µM, and 6 µM, respectively (Fig. 2a). Indeed, these five compounds are close structural analogs of each other, providing a 3CL protease inhibitor scaffold and structure-activity relationships for further optimization (Fig. 1c).

A The dose-dependent effects of DEL1 through DEL8 on the activity of 20 nM SARS-CoV-2 3CL protease were tested. The fluorogenic peptide MCA-AVLQSGFR-Lys(DNP)-Lys-NH2, corresponding to the nsp4/nsp5 cleavage site in the virus was applied as the substrate. Mean of 2 biological replicates is plotted. Source data are provided as a Source Data file. B Ability of DEL1 through DEL8 to inhibit SARS-CoV-2 viral infection in human Huh-7 cells, which were transduced to overexpress ACE2 receptor (Huh-7ACE2) before infection with SARS-CoV-2 virus (2019-nCoV/USA-WA1/2020). Data are plotted as the mean ± s.d., n = 3 biological replicates. Source data are provided as a Source Data file.
We evaluated these five compounds for inhibition of SARS-CoV-2 viral replication in human Huh-7 cells, which were transduced to stably overexpress the ACE2 receptor (Huh-7ACE2) before infection with a reporter SARS-CoV-2 virus (USA-WA1/2020 strain). We found that DEL2, DEL7, and DEL1 blocked viral infection, with EC50 values of 2 μM, 3 μM, and 4 μM, respectively (Fig. 2b). DEL3 and DEL8 also suppressed viral replication but exhibited EC50 values above 10 µM.
Co-crystal structures of DEL2 and DEL7 with SARS-CoV-2 3CL protease suggest non-covalent binding to the active site
Aiming to directly visualize the binding mechanism of top screening hits to 3CL protease, as well as to guide structure-based optimization efforts, we determined crystal structures of 3CL protease with DEL2 and DEL7 at resolutions of 1.85 Å and 1.74 Å, respectively (Fig. 3a, b, Supplementary Fig. 2a, Supplementary Table 1, 2). The X-ray crystal structures revealed that both compounds bind non-covalently to the substrate-binding pocket of 3CL protease. Since DEL7 is a close analog of DEL2, differing only by a fluoro substitution at the para position of the phenyl group, the two compounds share similar binding modes to the 3CL protease. Primary amines on both compounds strongly interact with the catalytic C145 residue. In both structures, the benzonitrile group occupies the S1 site, anchored by a hydrogen bond with the side chains of H163 (Fig. 3a, c, Supplementary Fig. 2b).

A The co-crystal structures of 3CL-DEL7 (yellow) complex. The inhibitor-interacting residues of 3CL protease subunit A (cyan) and subunit B (green, the other protomer of the 3CL homodimer) are shown with stick models. The Fo-Fc electron density omit map at 3σ is shown with blue mesh and hydrogen bonds are represented by red dash lines. C145 appeared to be oxidized to sulfenic acid during crystal growth and interacted with the primary amine of DEL-7. B Summary of atomic distances of hydrogen bonds between 3CL protease and DEL7 or DEL7 analogs in the co-crystal structures. C The structural overlay of 3CL protease bound to DEL7 with 3CL protease bound to GC376 (PDB code: 7JSU), compound 4 (PDB code: 7JT7), and nirmatrelvir (PDB code: 7U28). Substrate binding sites are labeled. The black arrow highlights how the peptidic backbones of GC376, compound 4, and nirmatrelvir overlaps well with each other, while the peptidic backbone of DEL7 and its analogs is flipped by almost 140° as compared with those of g-lactam-containing inhibitors. The depicted electrostatic surface (KbT/e = ±5.0) of 3CL protease alone (PDB code: 7JST) was generated using the program APBS42 and rendered in PyMOL. Clashes of inhibitors with the surfaces demonstrated how inhibitors re-organized the substrate-binding site and its plasticity. D MALDI-MS spectra of 3CL only (gray), 3CL treated with 10 equivalent (eq) DEL7 for 1 h at 4 °C (blue), 3CL preincubated with DMSO for 1 h at 4 °C before treatment of 10 eq Compound 4 for 1 h at 4 °C (green), 3CL pre-incubated with 1 eq GC376 for 1 h at 4 °C before treatment of 10 eq Compound 4 for 1 h at 4 °C (red), and 3CL preincubated with 1 eq DEL7 for 1 h at 4 °C before treatment of 10 eq Compound 4 for 1 h at 4 °C (purple).
The dichlorophenyl group of both compounds is favorably embedded in the hydrophobic S2 site, surrounded by the alkyl portion of the side chains of H41, M49, M165, D187, and Q189. More importantly, the dichlorobenzyl group forms methionine-aromatic motifs15 (sulfur-π interactions) with both M49 and M165, which were found to yield additional stabilization as compared with purely hydrophobic interaction via isobutyl groups commonly seen in the S2 site with previously reported 3CL inhibitors7,13 (Fig. 3c, Supplementary Fig. 2c).
The p-Cl group establishes either a hydrogen bond or hydrophobic interactions with the side chain of H41. Extending into the S3 and S4 sites, the NH of the terminal amide bond of DEL2 and DEL7 is stabilized by a hydrogen bond with the side chain of E166. However, we observed stronger electron densities, which typically indicate a more specific recognition and tighter binding, in the S3 and S4 sites for DEL7, primarily due to the formation of a hydrogen bond between the p-F group and the NH backbone of A191.
The crystal structure of the 3CL protease with DEL7 revealed a strong, continuous electron density map between the primary amine and the thiol of the catalytic C145 residue. Considering the implausibility of a covalent interaction between these two groups, this suggested that Cys145 was oxidized to a sulfenic acid in the course of crystal growth. Such oxidation of cysteine thiols has been observed at acidic pH with neighboring histidine as a proton acceptor, which is in line with our crystallization conditions (pH = 5–6) and the presence of H41 in the active site. Indeed, the presence of an oxygen atom bridging a sulfur and nitrogen atom to form the S-O-N entity is a widespread occurrence16.
To complement this crystallographic study, we further investigated the binding mode of DEL7 by mass spectrometry (MS). In a Matrix-Assisted Laser Desorption Ionization (MALDI) MS analysis of the intact 3CL protease protein after incubating it with DEL7 or covalent inhibitor Compound 4, we observed covalent modification only by the control Compound 4, with a mass shift matching its molecular weight (Fig. 3d). No mass shift induced by DEL7 was observed, which was consistent with a non-covalent binding mechanism of DEL7 to the 3CL protease.
In prior studies, we found that the reversible covalent inhibitor GC376 would not shift the mass of 3CL in MALDI MS due to the sensitivity of its covalent bond to pH changes, but pre-incubation of the 3CL protease with one equivalent of GC376 would occupy the active site and block the subsequent binding of Compound 4 to 3CL protease. Accordingly, we pre-incubated 3CL protease with one equivalent of DEL7 or GC376 before the addition of Compound 4. While GC376 consistently blocked Compound 4 binding through a reversible covalent interaction, DEL7 exhibited no such effect (Fig. 3d). Together, these data support a non-covalent binding mechanism of DEL7 to the SARS-CoV-2 3CL protease, as expected based on its chemical structure lacking an electrophilic warhead and several other crystal structures that we determined with DEL7 analogs.
DEL7 and its analogs specifically inhibit coronavirus 3CL proteases
Selectivity is considered a major challenge for targeting SARS-CoV-2 proteases for the treatment of COVID-19, as reported 3CL protease inhibitors, including GC376, are also potent inhibitors of human cathepsin L and B17. Cathepsin L was shown to mediate SARS-CoV-2 viral entry, but the virus may adopt alternative entry pathways, such as TMPRSS2, a transmembrane serine protease18,19. Therefore, dual inhibitors of 3CL protease and cathepsin L exhibit a partial loss of their antiviral activities in cells expressing TMPRSS2, which limits their clinical usefulness17. This selectivity limitation may be especially relevant for repurposing inhibitors of other proteases for 3CL protease or applying compounds with covalent warheads designed to target cysteine proteases.
By comparison, DEL7 and its analogs are non-covalent inhibitors identified from targeted screening specifically for the SARS-CoV-2 3CL protease. In line with this, we found that none of these hits inhibited human cathepsin L at concentrations up to 10 µM (Supplementary Fig. 3a), while GC376 exhibited an IC50 value of 0.33 nM on cathepsin L.
We further evaluated DEL7 against a panel of additional human proteases of various classes (Supplementary Fig. 3b). The result confirmed that DEL7 is highly selective for 3CL inhibition, displaying IC50 values of >10 μM against all tested human proteases. For therapeutic consideration, we measured the cellular toxicity of all DEL7 analogs in human cells, and no compound exhibited toxicity at concentrations up to 10 μM (Supplementary Fig. 4).
Considering that the 3CL protease substrate-binding pocket is highly similar across 12 different coronaviruses, we hypothesized that small-molecule inhibitors of the SARS-CoV-2 3CL protease have the potential to be broadly effective against other coronaviruses5,6. To test this hypothesis, we transfected HEK293T cells with 3CL proteases from different coronaviruses to induce cytotoxicity so that inhibitors of the corresponding 3CL protease could protect cells20. Accordingly, we found that DEL7 not only exhibited inhibitory effect on the SARS-CoV-2 3CL protease in cells, but also suppressed the 3CL protease activity of SARS-CoV and MERS-CoV in cells (Supplementary Fig. 3c). Moreover, this cell-based assay confirmed that DEL7 can effectively inhibit the tested viral proteases within a cellular context, in agreement with the expected mechanism of its antiviral activity.
Structural optimization of DEL7 led to the discovery of analogs with improved biochemical and biophysical potency for SARS-CoV-2 3CL protease
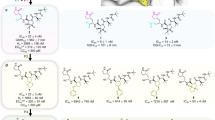
We focused on the creation of improved analogs of DEL7, based on its highly specific binding and strong electron density in the co-crystal structure. To optimize the structure of DEL7, we used a structure-based approach. We defined five modifiable groups in the chemical structure of DEL7, termed R1, R2, R3, R4, and R5 (Fig. 4). In the design of the DEL7 analogs, we modified one group at a time.

Structures of DEL7 analogs, along with their IC50 values against SARS-CoV-2 3CL protease activity in a biochemical assay and EC50 values against SARS-CoV-2 viral replication in a Huh7ACE2 cellular viral assay. Structure differences from DEL7 are shown in red color.
Since the R1 group has weaker electron density than other groups in the co-crystal structure of DEL7 with the 3CL protease, more analogs with modifications on R1 were synthesized initially (Fig. 4). Compared with DEL7 in the biochemical assay, two analogs featured improved inhibition potency on SARS-CoV-2 3CL protease: 1 and 2, with IC50 values of 70 nM and 20 nM, respectively. Structurally, both analogs have an elongated alkyl moiety with a terminal dimethyl amino group, where 2 features one additional methylene group in the alkyl chain compared with 1. Other analogs (3–9) with modifications on R1 exhibited similar or lower potency in comparison with DEL7.
DEL2 (Fig. 4) is a close analog of DEL7, differing solely by the fluorine on the R2 phenyl group. It exhibited comparable biochemical potency to DEL7, prompting the testing of fluorine removal on additional optimized DEL7 analogs.
For R3, we substituted both chlorines in DEL7 with fluorines and generated 10, which exhibited a poorer biochemical potency than DEL7, with an IC50 value of 2 µM. This may be due to the loss of hydrophobic interactions or a hydrogen bond interaction between the p-Cl in the R3 group of DEL7 and the side chain of H41. For the R4 group, which resides next to the unoccupied S1’ site in the co-crystal structure of DEL7 with 3CL protease, we added acetyl or tert-butyloxycarbonyl (Boc) groups onto the amine, aiming to occupy the S1’ site to establish more favorable interactions. The resulting analogs 11 and 12 lost inhibitory potency in the biochemical assay, suggesting the importance of interactions between the primary amine in the R4 group of DEL7 and C145.
DEL1 (Fig. 4) is also a close analog of DEL7, the only difference being the substitution of the nitrile moiety in R5 with a nitro group. Additionally, the R5 group occupies the S1 site, where a γ-lactam group has commonly been designed in previously reported 3CL protease inhibitors, including GC37613, compound 414, and nirmatrelvir7. We therefore substituted the R5 group of DEL7 with a γ-lactam and generated 13. However, both DEL1 and 13 were less active than DEL7 in the biochemical assay, suggesting that benzonitrile is preferred in the R5 group in the context of the DEL7 scaffold.
The structural overlay of 3CL protease bound to DEL7 with 3CL protease bound to GC376, compound 4, or nirmatrelvir provides a tantalizing clue to why the lactam is not a preferred moiety for DEL7: the peptidic backbone of DEL7 and its analogs is flipped by almost 140° as compared with those of γ-lactam-containing inhibitors (Fig. 3c, Supplementary Fig. 2c).
We further evaluated the binding of DEL7 and its improved analogs 1 and 2 to the 3CL protease in a surface plasmon resonance (SPR) assay and observed KD (dissociation constant) values of 434 nM, 196 nM, and 63 nM, respectively (Fig. 5a). The improved biophysical binding affinities observed for 1 and 2 are in line with their improved biochemical potencies in the 3CL protease assay.

A Surface plasmon resonance (SPR) analysis of the in vitro binding of DEL7, 1, and 2 to SARS-CoV-2 3CL protease. Different concentrations of inhibitors were serially injected onto a sensor chip with immobilized 3CL protease. The equilibrium dissociation constant (Kd) of the inhibitors was obtained by fitting binding response units to the Hill equation. B, C The co-crystal structures of 3CL-1 (purple) and 3CL-2 (orange) complex. The inhibitor-interacting residues of 3CL protease subunit A (cyan) and subunit B (green, the other protomer of the 3CL homodimer) are shown with stick models. The Fo-Fc electron density omit map at 3σ is shown with blue mesh. Hydrogen bonds and hydrophobic interactions are represented by red and blue dash lines, respectively. In panel (C), C145 appeared to be oxidized to sulfenic acid during crystal growth, but it didn’t form a covalent bond with the primary amine of DEL7-5. D Metabolic stability of DEL7 and 2 in human and mouse (CD-1) liver microsomes.
Aiming to decipher the basis of improvement over DEL7 as well as to guide further optimization efforts, we solved the X-ray crystal structures of 3CL protease with 1 and 2 at resolutions of 1.93 Å and 1.56 Å, respectively (Fig. 5b, c, Supplementary Table 3, 4). While both compounds inherited the interactions that DEL7 featured with the protease, the elongated alkyl chains in the R1 group further extended beyond the S3 site, reaching toward the other protomer (B) of the physiological homodimer of 3CL protease. Specifically, the tertiary amine in the R1 group of 2 was anchored by additional hydrophobic interactions with S1 and N214 of protomer B, which may explain its improvement over DEL7. These interactions were absent in previously reported γ-lactam-containing 3CL inhibitors (Fig. 3c). Moreover, we found that the C145 of the 3CL protease co-crystalized with 1 had been oxidized to sulfenic acid (Fig. 5b). This observation confirmed that the continuous electron density map between the DEL7 primary amine group and the thiol group of C145 is an oxygen atom bridging a sulfur and nitrogen atom to form the S-O-N entity due to crystallographic artifacts introduced during crystal growth at pH 5-616.
For consideration of therapeutic applications, we evaluated the metabolic stability of DEL7 and 2 in human and mouse liver microsomes and observed a significant improvement of 2 (T1/2, human = 7.6 min, T1/2, mouse = 41 min) over DEL7 (T1/2, human = 1 min, T1/2, mouse = 0.9 min), indicating potential suitability for further animal studies and an approach to enhance the metabolic stability of DEL7 via modifications of the R1 group (Fig. 5d).
Cell permeability limits antiviral potencies of DEL7 analogs but can be overcome by reducing net charge
Based on the improved biochemical potency observed for 2 over 1 and DEL7, we continued modifying the R1 group in DEL7. We elongated the alkyl chain in 2 by adding one or two methylene groups and generated 14 and 15, respectively (Fig. 6a). We also elongated the R1 group of 2 by incorporating the nitrogen-containing heterocycles triazole or piperazine and generated 16 and 17, respectively (Fig. 6a).

A Structures of DEL7 analog 14, 15, 16, and 17, along with their IC50 values against SARS-CoV-2 3CL protease activity in a biochemical assay and EC50 values against SARS-CoV-2 viral replication in a Huh7ACE2 cellular viral assay. B–E The co-crystal structure of 3CL-14 (brown), 3CL-15 (magenta), 3CL-16 (pink), and 3CL-17 (violet) complex. The inhibitor-interacting residues of 3CL protease subunit A (cyan), subunit B (green, the other protomer of the 3CL homodimer), and subunit C (pink, a neighboring protomer in the crystal packing, not part of the 3CL homodimer formed by subunit A and B) are shown with stick models. The Fo-Fc electron density omit map at 3σ is shown with blue mesh. Hydrogen bonds, hydrophobic interactions, and methionine-aromatic motif15 are represented by red, blue, and green dash lines, respectively. The methionine-aromatic motif (Sulfur-π interactions) was found to yield additional stabilization as compared with purely hydrophobic interaction15. In panel E, C145 appeared to be oxidized to a sulfenic acid during crystal growth, but it didn’t form a covalent bond with the primary amine of 17.
When tested against the SARS-CoV-2 3CL protease, 14 and 15 exhibited further improvements over 2. Both 14 and 15 featured IC50 values of 10 nM, the highest possible potency in the biochemical assay, which uses 20 nM protease to cleave enough fluorogenic substrates to produce a fluorescence signal detectable by our instrument. Analogs 16 and 17 were also able to efficiently inhibit 3CL protease, with IC50 values of 0.5 µM and 30 nM, respectively. This validated the strategy of modifying the R1 group for further optimization.
Aiming to decipher the basis for these improvements, as well as to guide further optimization efforts, we determined the X-ray crystal structures of the 3CL protease with 14, 15, 16, and 17, at resolutions of 1.55 Å, 1.67 Å, 1.51 Å, and 1.72 Å, respectively (Fig. 6b–e, Supplementary Table 5, 6, 7, 8). Compared with the co-crystal structure of 2, these four compounds retained interactions that 2 featured with the active site of the protease, including the hydrogen bonds with H163, E166, and A191 (Fig. 6b). Furthermore, the R1 groups of these compounds were engaged in and stabilized by additional interactions with the active site and the interacting protomer B of the native 3CL protease homodimer. Specifically, the R1 group of 15 recruited the penultimate residue F305 of protomer B into hydrophobic interactions (Fig. 6c). The R1 group of 16 was held tightly by two additional hydrogen bonds engaging the side chain of H64 from a neighboring protease protomer (C) and the backbone carbonyl of P168, in addition to a π-π interaction between the triazole ring of 16 and F305 of protomer B (Fig. 6d). In contrast, the piperazine nitrogen of 17 forms a hydrogen bond with the backbone carbonyl of N214 of protomer B (Fig. 6e). These additional interactions stabilize the R1 group of these compounds in the substrate binding site and on the surface of the other protomer of 3CL homodimer, as evidenced by strong and well-defined electron densities in the crystal structures. This is in line with their high potency in biochemical assays.
However, when tested in cells infected with SARS-CoV-2, 2 did not show improvement over DEL7. Indeed, its EC50 value (4.2 µM) was poorer than that of DEL7 (3.3 µM). Analogs 1, 14, 15, and 17 only exhibited slight improvements over DEL7 in this cellular viral assay, with EC50 values of 1.2 µM, 1.3 µM, 1.7 µM, and 0.8 µM, respectively. However, their EC50/IC50 ratios (1.2/0.07 = 17, 1.3/0.01 = 130, 1.7/0.01 = 170, 0.8/0.03 = 27, respectively) were markedly higher than for DEL7 (3.3/0.3 = 11). We speculated that this poor cellular potency might be due to stability or permeability issues. To investigate how we could resolve this issue by optimizing the structure, we synthesized seven additional analogs of the most biochemically potent compound, 14, with modifications on R1 through R5 (Fig. 7).

Structures of DEL7 analogs designed on top of analog 14, along with their IC50 values against SARS-CoV-2 3CL protease activity in a biochemical assay and EC50 values against SARS-CoV-2 viral replication in a Huh7ACE2 cellular viral assay. Structure differences from DEL7 are shown in red color.
We first shortened the linker to R1 by one methylene and generated 18 (Fig. 7); this caused a complete loss of potency. This is likely due to the loss of a critical hydrogen bond between the R1 amide NH and the E166 side chain. We then replaced the fluorine in the R2 group with hydrogen and prepared the DEL2-like compound 19, as DEL-2 featured a better EC50/IC50 ratio (2/0.3 = 7) than DEL7. However, this trend was not reproduced in the case of 19, as it showed an EC50/IC50 ratio of 103 (3.1/0.03). For R3, we removed either of the chloro groups from the dichlorobenzene to decrease the molecular weight of the compounds. However, the resulting analogs 20 and 21 exhibited much lower potency than 14 in both biochemical and cellular assays, indicating that both chlorines are important for potency. Moreover, all crystal structures complexed with DEL compounds confirmed this finding, as p-Cl either forms polar or non-polar interactions with H41 and D87, while o-Cl interacts with the F-benzyl group of each inhibitor.
We then modified the chirality of the (R)-amino group in R4 by generating a diastereomer 22 with (S)-NH2. We observed a substantial loss of inhibition potency, suggesting a specific stereochemistry requirement for the primary amine.
We found that removal of the R4 amino group was tolerated, as the corresponding analog 23 (R4 = H) exhibited an IC50 value of 0.07 µM and an EC50 value of 2.3 µM. Interestingly, a better EC50/IC50 ratio (2.3/0.07 = 33) was observed for 23. Compared with 14, which carries two positive charges at physiological pH due to the primary amine in R4 and the tertiary amine in R1, 23 only carries one positive charge. Highly charged small molecules are typically less cell permeable, so removal of charge from DEL7 analogs appeared to be a feasible approach to improve cellular permeability, as well as cellular antiviral potency. This is evidenced by the undesirably high EC50/IC50 ratio observed on highly charged DEL7 analogs, including 2 (4.2/0.02 = 210), 15 (1.7/0.01 = 170), and 19 (3.1/0.03 = 103).
We also replaced the nitrile group in R5 of 14 with an amide group, but it caused a substantial decrease of inhibition potency (24, IC50 = 0.3 µM).
Aiming to validate the strategy of restricting net charge in DEL7 structural optimization, we subsequently modified R1 with neutral groups, as modification of R1 was shown to be more efficient in improving DEL7 as compared to other sites. We, therefore, synthesized 25 and 26 (Fig. 8a), elongating the alkyl chain of DEL7 R1 group by three methylene groups in 26, and incorporating an alkyne in the R1 group of 25. Compared with DEL7 (IC50 = 0.3 µM, EC50 = 3.3 µM) in the biochemical and cellular assay, 25 (IC50 = 0.1 µM, EC50 = 0.9 µM) and 26 (IC50 = 0.2 µM, EC50 = 0.8 µM) exhibited moderate improvements.

A Structures of DEL7 analog 25 and 26, along with their IC50 values against SARS-CoV-2 3CL protease activity in a biochemical assay and EC50 values against SARS-CoV-2 viral replication in a Huh7ACE2 cellular viral assay. Structure differences from DEL7 are shown in red color. B The co-crystal structure of 25 (dark purple) complex. The inhibitor-interacting residues of 3CL protease subunit A (cyan) and subunit B (green, the other protomer of the 3CL homodimer) are shown with stick models. The Fo-Fc electron density omit map at 3σ is shown with blue mesh. Hydrogen bonds and hydrophobic interactions are represented by red and blue dash lines, respectively. C Cytosolic concentrations of DEL7, 14, and 25 in HEK293T cells treated with 10 µM of each compound. Data are plotted as the mean ± s.d., n = 3 biological replicates. Source data are provided as a Source Data file.
When we further evaluated 25 for inhibition of HCoV-Alpha-229E and HCoV-Beta-OC43 viral infection in Huh7 or RD cells, we found that 25 effectively blocked viral infection of HCoV-Alpha-229E and HCoV-Beta-OC43 with EC50 values of 2.5 µM and 3.3 µM, respectively (Supplementary Table 9). The consistent activity of 25 on human coronaviruses in addition to SARS-CoV-2 (EC50 = 0.9 µM) suggested the potential of developing DEL7 scaffold into a pan-coronavirus inhibitor.
We determined a crystal structure of the 3CL protease with 25, at resolution 1.81 Å (Fig. 8b, Supplementary Table 10). The electron density corresponding to 25 is not the highest among the DEL7 analogs that we determined; its terminal methyl group does not show well-defined electron density, despite forming a hydrophobic interaction with Cα of G170. This is in line with its moderate potency in inhibiting the purified protease.
However, to test our hypothesis that cell permeability is the limiting factor for cellular potency, we treated 293 T cells with 10 µM DEL7, 14, or 25, and quantified each compound’s concentration in cells using LC-MS. The results suggested that 14, which is doubly charged, is much less permeable than DEL7 and 25, which carry only one positive charge at the primary amine (Fig. 8c). Thus, the alkyne in R1 on 25 allows high cell accumulation. This supports our hypothesis and confirms the strategy to optimize analogs with less net charge.
Structural optimization of DEL7 with restrictions on net charge led to discovery of analogs with improved antiviral potency for SARS-CoV-2
Aiming to optimize the DEL7 structure into a potent antiviral inhibitor in cellular context, we prioritized synthesizing DEL7 analogs with less net charge than 14. This strategy included modification of R1 with neutral groups and substitution of unnecessary charged groups on compounds exhibiting high potency. We therefore synthesized a panel of 25 additional analogs of DEL7 (Fig. 9a and Supplementary Figs. 5, 6).

A Structures of DEL7 analogs 27, 28, 29, 30, 31, 32, and 46, along with their IC50 values against SARS-CoV-2 3CL protease activity in a biochemical assay and EC50 values against SARS-CoV-2 viral replication in a Huh7ACE2 cellular viral assay. Structure differences from DEL7 are shown in red color. B–H, The co-crystal structures of 3CL-27 (navy), 3CL-28 (dark pink), 3CL-29 (dark green), 3CL-30 (chocolate), 3CL-31 (light green), 3CL-32 (salmon), and 3CL-46 (olive) complex. The inhibitor-interacting residues of 3CL protease subunit A (cyan), subunit B (green, the other protomer of the 3CL homodimer), and subunit C (pink, a neighboring protomer in the crystal packing, not part of the 3CL homodimer) are shown with stick models. The Fo-Fc electron density omit map at 3σ is shown with blue mesh. Hydrogen bonds and hydrophobic interactions are represented by red and blue dash lines, respectively. I, The structural overlay of 3CL protease bound to DEL7 with 3CL protease bound to 15 DEL7 analogs. The shown electrostatic surface (KbT/e = ±5.0) of 3CL-DEL7 complex was generated using program APBS42 and rendered in PyMOL.
Compared with DEL7 (IC50 = 0.3 µM, EC50 = 3.3 µM) in the biochemical and cellular assay, 27 (IC50 = 0.1 µM, EC50 = 0.6 µM), 28 (IC50 = 0.02 µM, EC50 = 0.7 µM), 29 (IC50 = 0.04 µM, EC50 = 0.3 µM), and 30 (IC50 = 0.05 µM, EC50 = 1.0 µM) exhibited substantial improvements (Fig. 9a). All these compounds feature modifications to their R1 groups.
While the alkyl chain of the 25 R1 group is elongated by two more methylene groups in 27, the tertiary amine of 14 was replaced by pyridine to generate 29 and 30, as pyridine is also nitrogen-containing but neutral at physiological pH (pKa = 5.2). Alternatively, we attempted to alter the charge of 14 by substituting its tertiary amine with pyrrolidine, a cyclic amine. The resulting analog 28 was still a potent 3CL inhibitor (IC50 = 0.02 µM) but its permeability was only slightly improved, as it exhibited an EC50/IC50 ratio of 35 (0.7/0.02), which is the highest among these four compounds. In contrast, when we modified R1 with neutral groups containing phenyl or ether moieties to generate 31 or 32, they exhibited the lowest EC50/IC50 ratios of all DEL7 analogs (0.3/0.5 = 0.6 and 0.4/0.3 = 1.3). Although 31 and 32 only feature moderate potency in inhibiting the purified protease (IC50 = 0.5 µM and 0.3 µM, respectively), they exhibited substantial improvements over DEL7 in the cellular antiviral assay. The correlations between net charge and EC50/IC50 ratio were also reflected in DEL7 analogs with other modifications of the R1 group, including imidazole (33), morpholine (34), nitrile (35 and 36), carboxylic acid (37), and sulfonyl (38 and 39), which further validated this strategy.
In addition to modifying the R1 group, we also attempted to remove the charged primary amine in the R4 site of 7, initially through masking it with acyl or sulfonyl groups. We observed a substantial loss of inhibition potency in the corresponding analogs 40, 41, 42, and 43. We tested replacement of the primary NH2 with neutral groups OH or Cl. Analogs with hydroxyl in place of amine, 44 and 45, were much less potent than 7 (IC50 = 0.01 µM) in inhibiting the purified protease (IC50 = 0.5 µM and 0.6 µM). However, we found that substitution of the amino group by chloro was tolerated, as the corresponding analog 46 exhibited an IC50 value of 0.01 µM; but it still exhibited a high EC50/IC50 ratio, suggesting modification of R1 is more efficient to develop analogs with improved cellular antiviral potency.
We determined crystal structures of 3CL protease with 27, 28, 29, 30, 31, 32, and 46, at resolution 1.90 Å, 1.71 Å, 1.52 Å, 1.71 Å, 1.55 Å, 1.57 Å, and 1.55 Å, respectively (Fig. 9b–i, Supplementary Table 11, 12, 13, 14, 15, 16, 17). Compared with the co-crystal structure with DEL7, we found that these compounds retained interactions that DEL7 featured with the active site of the protease, including the hydrogen bond or polar interactions with H41, H163, E166, and A191. Similar to what we observed with 14 and 15, the R1 groups of these compounds were engaged and stabilized by additional interactions with the active site and the interacting protomer B of the 3CL protease homodimer. Specifically, the alkyl chain in the R1 group of 46 was anchored by multiple hydrophobic interactions with G170 and surrounding side chains of residues from neighboring protomers. The pyridine in the R1 group of 29 engaged in π-π interactions with F305 of protomer B and H64 from a neighboring protease protomer (C). These additional interactions stabilized the R1 group of these compounds in the substrate binding site. The electron densities corresponding to 29 and 30 are among the highest of all DEL7 analogs that we determined. This is in line with their high potency in biochemical assays.
For consideration of therapeutic applications, we evaluated the metabolic stability of DEL7, 27, 29, and 32 in mouse liver microsomes and observed improvements of 27, 29, and 32 (T1/2 = 11 min, 2.5 min, and 14 min, respectively) over DEL7 (T1/2 = 0.9 min), indicating modifications of the R1 group can enhance both antiviral potency and the metabolic stability of DEL7 (Supplementary Table 18). When we further evaluated 29 for inhibition of viral replication of the Omicron variant of SARS-CoV-2 (XBB lineage, hCov-19/USA/CA-Stanford-109_S21/22 strain) in human Caco-2 cells, we found that 29 fully blocked viral infection of the Omicron variant, at concentrations as low as 0.1 µM (Supplementary Table 19).
To summarize the structural optimization of DEL7, we found that elongation of the alkyl chain of the R1 group with incorporation of nitrogen-containing groups allows extension beyond the S3 site, reaching towards the other protomer of the native homodimer of 3CL protease, engaging additional interactions with the active site and the interacting protomer, and substantially improving potency for inhibiting the purified protease (Supplementary Fig. 7). In addition, net charges on the R1 group should be eliminated to enhance cell permeability and cellular antiviral potency.
Combining both strategies, analog 29 was found to be a potent 3CL protease inhibitor that can efficiently block SARS-CoV-2 replication in cells, so it may serve as a development candidate for treating COVID-19 and other coronavirus infections. We have termed this compound hermestat, after the Greek deity Hermes, who could rapidly move between worlds, reflecting the non-covalent nature of this compound.
Discussion
Since the outbreak of the COVID-19 pandemic, drug repurposing or re-optimization of known inhibitors has been broadly adopted to search for treatment. Disadvantages of drug repurposing range from dosing uncertainty and safety concerns to potential side effects and limited effectiveness. Re-optimization of known inhibitors may be constrained by specific scaffolds and lead to similar binding mechanisms and resistance. More importantly, human cells contain 26 selenocysteine proteins trafficking in different compartments of the cell, particularly membranes. There is a poor understanding of their specific functions, as structural information for most of them is unavailable21. For such proteins with known functions, there is substantial evidence of their roles in human diseases. As selenocysteine is more reactive than cysteine towards electrophilic covalent inhibitors, how covalent inhibitors may inhibit or alter the functions of these selenoproteins has not been investigated. Therefore, it is valuable to create non-covalent inhibitors with diverse scaffolds for viral proteases.
Here, we started a de novo screen to search for non-covalent inhibitors specifically targeting SARS-CoV-2 3CL protease. The top screening hit was selected from billions of structurally diverse compounds in a DNA-encoded chemical library (DEL). Distinct from the covalent 3CL inhibitor nirmatrelvir, these compounds are non-covalent inhibitors, which feature different and potentially more favorable profiles. Specifically, optimized analog 29 is among the top compounds in comparison with the current benchmark SARS-CoV-2 3CL protease non-covalent inhibitors (Table 1) and featured a binding mode that is distinct from other non-covalent inhibitors9,22,23,24,25,26,27,28,29 (Supplementary Fig. 8). In addition, as direct analogs of the screening hit DEL7, these compounds inherited the binding mode difference that DEL7 exhibited in comparison with nirmatrelvir (Fig. 3c, Supplementary Fig. 2c, 8h). They may, therefore, overcome resistance and rebound issues reported for nirmatrelvir and complement its clinical utility.
The optimized DEL7 analogs that we developed are capable of occupying the substrate-binding site. It is noteworthy that the superposition of the 12 crystal structures of 3CL proteases from different coronaviruses shows that the substrate-binding site is highly conserved among 3CL protease in all coronaviruses5,6. Therefore, optimized DEL7 analogs have the potential to be broadly effective against emerging variants of SARS-CoV-2 or other coronaviruses.
Furthermore, optimized DEL7 analogs with elongated R1 groups were shown to interact with the other protomer of the 3CL native homodimer. This specific binding mode of optimized DEL7 analogs to the 3CL protease is different from all other current benchmark SARS-CoV-2 3CL protease non-covalent inhibitors, as demonstrated by the clear difference of 29 in a structural overlay of 3CL bound to it and other non-covalent inhibitors (Supplementary Fig. 8). It has been reported that only dimeric 3CL protease is catalytically competent30,31,32. This is in line with our observation of improved potency in inhibiting purified protease when we elongated the R1 group of DEL7 to engage interactions with the other 3CL protomer. Furthermore, we found that the longer the alkyl chain of R1 group is, the better its inhibition potency is. This was observed on the series of analog 1 (two methylene groups in the alkyl chain, IC50 = 0.07 µM), 2 (three methylene groups in the alkyl chain, IC50 = 0.02 µM), 14 (four methylene groups in the alkyl chain, IC50 = 0.01 µM), and 15 (five methylene groups in the alkyl chain, IC50 = 0.01 µM).
This trend was also observed in the series of 3 (zero methylene group in the alkyl chain, IC50 = 8 µM) and 4 (two methylene groups in the alkyl chain, IC50 = 0.6 µM). Analogs 35 (one methylene group in the alkyl chain, IC50 = 0.6 µM) and 36 (two methylene groups in the alkyl chain, IC50 = 0.1 µM) exhibited this trend as well. Together, these data suggest interactions with the other protomer contributes to the improvement in inhibition potency. This finding also reveals insights into the design of drug-like inhibitors targeting both protomers of the native homodimer of 3CL protease, including length of alkyl chain as a linker and functional groups that can perturb both protomers. Thus, the optimized compound hermestat and the insights obtained in its development may lead to a class of pan-coronavirus, non-covalent therapeutics that will be valuable for treating current and future coronavirus infections.
Optimization started from DEL7, a hit from DEL screening. The concept of DEL screening was reported 30 years ago, but was re-initiated and expanded in the last decade, thanks to the advances in high-throughput sequencing techniques33. DEL screening requires no special instruments. Furthermore, parallel DEL selections can be easily conducted, making it possible to directly compare results across different conditions. This could conveniently enable selection of variant-specific binders, or binders that feature different or similar binding sites to known inhibitors. Indeed, our application of DEL screening was designed to discover small molecules that bind to the highly conserved substrate-binding site, which may increase the chance of discovering pan-coronavirus active inhibitors. In addition, since strong binders need to be eluted from the target protein before decoding the DNA label, a canonical DEL screening is suitable for identifying non-covalently reversible binding compounds. This is in line with our specific aim to develop non-covalent inhibitors for coronavirus proteases. It is noteworthy that WU-04 is another potent non-covalent 3CL protease inhibitor that was also identified from a DEL screening22 (Table 1). The structure and binding mode to 3CL protease of WU-04 are distinct from 29 (Supplementary Figs. 8, 9). Meanwhile, DELs that contain covalent warheads have been recently developed for discovery of covalent inhibitors of target proteins as well34. Together, these studies provide a roadmap to accelerate the discovery of treatments for future pandemic viruses using DNA-encoded library screening and structure-based compound optimization for viral proteases.
Methods
Preparation and synthesis of compounds
GC376 was purchased from Aobious. DNA-encoded chemical library (DELopen) screening hits (DEL1 - DEL8) were purchased from Wuxi AppTec, the provider of the DELopen library. Selected DEL7 analogs (2, 5, and 10) were purchased from chemical vendor Curia. Synthesis of all other compounds were summarized in the Supplemental Note 1.
Cell line
HT1080, HEK293T, PANC-1, Caco-2, and RD cells were acquired from ATCC with ATCC catalog number CCL-121, CRL-3216, CRL-1469, HTB-37, and CCL-136, respectively. Huh7 cell line is a gift from Matthew Evans, Mount Sinai. Cells were maintained at 37 °C in a humidified atmosphere with 5% CO2. Cells were confirmed mycoplasma negative prior to use.
Expression and purification of SARS-CoV-2 3CL protease
SARS-CoV-2 3CL protease with no tag or a C-terminal 6xHis tag was expressed and purified as previously described4. To utilize Ni-NTA magnetic beads to pull down 3CL protease and small molecule binders in the DEL screening, we added the 6xHis tag to the C-terminus of the previously constructed bacterial expression vector (pGEX-5X-3-SARS-CoV-2-3CL, Addgene 168457) either directly (e.g., VTFQHHHHHH) or following a Gly-Pro linker (e.g., VTFQGPHHHHHH) or Gly-Gly-Gly-Ser linker (e.g. VTFQGGGSHHHHHH) by PCR and Gibson Assembly. We designed these three different His-tag constructs (with or without linkers) so that we can test for the most efficient immobilization on Ni-NTA magnetic beads and use that construct for the screening. Although these constructs contain His-tagged, we constantly utilized the GST tag for purification of recombinant protein as we did on 3CL protein without His-tag. After confirmation by Sanger sequencing, the constructs were transformed into BL21 (DE3) cells. Briefly, these cells were grown at 37 °C, 220 RPM until OD 0.6-0.7, and then induced with 0.5 mM IPTG and further incubated at 16 °C, 180 RPM for 10 h. Cells were then pelleted, resuspended in lysis buffer (20 mM Tris-HCl, pH 8.0, 300 mM NaCl), sonicated, then ultracentrifuged at 25,000 × g for 1 h at 4 °C. This supernatant was incubated with Glutathione Sepharose resin (Sigma, GE17-5132-01) for 2 h at 4 °C, washed ten times with lysis buffer, then Factor Xa was added and further incubated for 18 h at 4 °C. Finally, the supernatant was collected, concentrated, and loaded onto a Superdex 10/300 GL column. Appropriate fractions were pooled and concentrated with a 10 kDa centrifugal concentrator (Amicon® Ultra Centrifugal Filter, Millipore UFC9010). Recombinant protein concentration was determined by Pierce BCA protein Assay Kit (Thermo Fisher, 23225) with BSA standards.
DNA-encoded chemical library (DELopen) screening for 3CL binders
Access to DELopen, a DNA-encoded library of 2.8 billion compounds (as of the screening date), was provided by Wuxi Apptec. We performed the screen against 3CL using the DELopen kit and following the recommended protocol. To capture 3CL protein onto beads for a pull-down binding affinity screening, we used His-tagged 3CL and Ni-NTA magnetic beads. Aiming for an efficient immobilization, we expressed and purified three types of His-tagged 3CL protein: 3CL-6xHis with no linker, 3CL-GP-6xHis (GP works as a linker between 3CL protein and His tag), and 3CL-GGGS-6xHis. The incorporation of the 6xHis tag was confirmed by Western Blot with 6x-His Tag Monoclonal Antibody (1:1000 dilution, Invitrogen, 4E3D10H2/E3, Mouse Monoclonal, Catalog # MA1-135, RRID: AB_2536841). A bead capture test was performed to test protein-beads immobilization efficiency. For each protein, 25 µL HisPur™ Ni-NTA magnetic beads (Thermo Scientific, #PI88831) were washed with 200 µL Washing Buffer (WB, provided by DELopen kit) for three times, using a magnetic rack (DynaMag magnet, Invitrogen, #12321D) to separate beads from the supernatant. Beads were then suspended with 125 µL Selection Buffer (SB, provided by DELopen kit), with 25 µL suspended beads collected as Blank Beads control. 6 µg His-tagged protein was dissolved in 120 µL SB, with 1 µg of each collected as Input control. The remaining 100 µL beads were pulled down, before removal of supernatant and mixing with the remaining 5 µg His-tagged protein in 100 µL SB. After an incubation for 30 min at 25 °C with gentle rocking, beads were pulled down with the magnetic rack, with supernatant collected as flowthrough. Beads were then washed with 100 µL SB and pulled down with the magnetic rack, with supernatant collected as Wash. After resuspension in 100 µL SB, 50 µL was collected as Beads, while the other 50 µL was incubated at 95 °C for 10 min. The heated beads were pulled down and supernatant was collected as Heated Elution. Eventually, the remaining beads were resuspended in 50 µL SB and collected as Heated Beads. The samples collected above were analyzed with gel electrophoresis and stained with Coomassie Blue before imaging to confirm protein-beads immobilization efficiency. 3CL-GP-6xHis was eventually used for immobilization onto beads before screening as we observed the most efficient immobilization with it on the beads.
In total, we used two DELopen kits. Since each kit provided four copies of library, to screen for 3CL specific binders, for the first kit, three copies of the library were separately screened against beads with 3CL protein as biological triplicates while the fourth copy of the library was screened against the negative control beads with no protein immobilized. For the second DELopen kit, four copies of the library were separately screened against beads capturing 3CL alone, 3CL-GC376 complex, 3CL-Compound 4 complex, or no protein as negative control. Such screening against protein alone and protein-inhibitor complex may enable identification of protein binders with same or different binding modes as the known inhibitors. This is because binders that share the same binding site as the known inhibitor will bind less efficiently on the protein-inhibitor complex than the protein alone. In contrast, binders with different binding sites from the known inhibitor will bind to the protein-inhibitor complex and the protein alone in an indistinguishable manner. Protein-inhibitor complex was prepared by incubating 50 μM of the 3CL-GP-6xHis with 500 μM of inhibitor in a buffer comprised of 50 mM Tris-HCl (pH 7.5), 1 mM EDTA for 1 h at 4 °C before immobilization onto beads.
Immobilization of His-tagged 3CL or 3CL-inhibitor complex was performed as described above for the beads capture test, using 20 µL beads and 5 µg protein as materials for each selection in each round. To include negative controls, protein buffer was used instead of protein to prepare vehicle control beads.
Three consecutive rounds of affinity selection were then performed. In each round of affinity selection, the beads for each corresponding condition were mixed with the library solution as dissolved in 100 µL SB. After an incubation for 1 h at 25 °C with gentle rocking, the beads were pulled down and washed with 200 µL SB buffer for three times to remove unbound library molecules. The library molecules bound on immobilized 3CL were released by incubating the SB-resuspended bead at 95 °C for 10 min, before pulling down the beads and collecting the supernatant. Part of the supernatant was set aside for future sequencing analysis while the remaining supernatant was diluted to 100 µL with SB and used as library solution for the next round of affinity selection. After the third selection, all heated supernatants were collected as elution for future sequencing analysis.
The samples acquired from affinity selection were returned to Wuxi AppTec, who performed sequencing to decode the DNA tags on library compounds to identify top binders. Wuxi AppTec then conducted the off-tag resynthesis for desired 3CL binders, sent us the resynthesized tag-free compounds for validation, and revealed the structure information for validated hits.
Measurement of SARS-CoV-2 3CL protease inhibition
The in vitro biochemical activity of the SARS-CoV-2 3CL protease was measured as previously described5: Practically, the fluorogenic peptide MCA-AVLQ/SGFR-Lys(DNP)-Lys-NH2, corresponding to the nsp4/nsp5 cleavage site in the virus, was synthesized (GL Biochem), then resuspended in DMSO to use as the substrate (slash (/) indicates the position where the proteolytic cleavage occurs).
For the measurement of IC50 values, in a 96-well plate, serial dilutions of the test compound were first prepared in the 3CL assay buffer (50 mM Tris-HCl, pH 7.5, 1 mM EDTA). The protease was subsequently added to each well at a final concentration of 20 nM. The substrate was added at 5 µM, and then fluorescence (Excitation 320 nm, Emission 405 nm) at 30 °C was continuously measured on a plate reader (Tecan Infinite F200, with the Tecan iControl software v.1.10) for 10 min. Inhibition was then calculated by comparison to control wells with no inhibitor added (negative control) and with no protease added (positive control). IC50 values were determined by fitting data with Log(inhibitor) vs. normalized response curve (the standard inhibition curve) in GraphPad Prism v10.0.
Chymotrypsin activity assay
Chymotrypsin Assay Kit (Abcam, ab234051) was applied to measure chymotrypsin activity. Panc-1 cells, which originated from human pancreas where chymotrypsin is synthesized, were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen, 11965092) supplemented with 10% Fetal Bovine Serum (FBS, Gibco, A4736201) and penicillin-streptomycin (PS, Invitrogen, 15140122), collected, and lysed on ice with the chymotrypsin assay buffer provided by the kit. Pierce BCA protein assay kit (Thermo Scientific, #23225) was applied to measure the total protein concentration of cell lysates. Lysates of 100 µg total protein was used for each assay on a 96-well plate and incubated with 10 µM of the test compound for 10 min on ice. Chymotrypsin activator and fluorogenic substrate were then added to make a total volume of 100 µL before fluorescence (Ex/Em = 380/460 nm) intensity was subsequentially recorded in a kinetic mode for 30 min at 25 °C. Inhibition was calculated by comparison to control wells with no compound added (negative control) and with no lysate added (positive control).
Caspase-3 activity assay
Caspase-3 Assay Kit (Abcam, ab39383) was used to measure caspase-3 activity. Casapse-3 of human HT1080 cells was activated by staurosporine (STS) using a previously optimized concentration and treatment time (1 µM STS for 9 h)35. 24 h before STS treatment, 5 million HT1080 cells were seeded in a 150-mm dish with DMEM supplemented with 10% FBS, non-essential amino acids (Invitrogen, 11140050), and PS. Cells were then treated with STS, collected, and lysed in the cell lysis buffer provided by the kit. Pierce BCA protein assay kit was applied to measure the total protein concentration of cell lysates. Lysates of 15 µg total protein was used for each assay on a 96-well plate and incubated with 10 µM of the test compound for 10 min on ice. Fluorogenic caspase-3 DEVD-AFC substrate were then added to make a total volume of 100 µL before fluorescence (Ex/Em = 400/505 nm) intensity was subsequentially recorded in a kinetic mode for 30 min at 25 °C. Inhibition was calculated by comparison to control wells with no compound added (negative control) and with no lysate added (positive control).
Human protease selectivity panel (including Cathepsin L)
Specific assay condition for each protease in our human protease selectivity panel is listed in Supplementary Table 20, including substrate, buffer, and fluorescence wavelengths. In general, protease was incubated with the test compound for 20 min before addition of fluorogenic substrate to initiate the reaction. Fluorescence intensity was then monitored over 2 h with 5 min interval. Inhibition was then calculated by comparison to control wells with no inhibitor added (negative control) and with no protease added (buffer only as positive control). Known inhibitor for each tested protease (listed in Supplementary Table 20) was included side-by-side to validate the assay. The slope of the linear portion of the fluorescence curve over time was calculated using Microsoft Excel (v16.35). IC50 values were determined by fitting an asymmetric sigmoidal curve to the data (GraphPad Prism, v10.0).
Mass spectrometry of protein-inhibitor complexes
To demonstrate the non-covalent binding mechanism of DEL7 to 3CL protease and exclude irreversible covalent binding, 50 μM 3CL protease was incubated with 500 μM DEL7 or DMSO (negative vehicle control) in the 3CL assay buffer for 1 h at 4 °C before mass spectrometry analysis. Irreversible covalent inhibitor Compound 4 was used as a positive control. To further exclude reversible covalent binding, 50 μM 3CL protease was first incubated with 50 μM DEL7 or DMSO (negative vehicle control) in the 3CL assay buffer for 1 h at 4 °C, before a complete buffer exchange to remove non-covalently bound inhibitor. Reversible covalent inhibitor GC376 was used as a positive control, which will not be removed by buffer exchange after covalent binding to 3CL. 500 µM of Compound 4 was then added to modify 3CL via an incubation for 1 h at 4 °C before mass spectrometry analysis.
Bruker ultrafleXtreme Matrix-Assisted Laser Desorption/Ionization Time-of-Flight (MALDI-TOF) was applied for mass spectrometry analysis. The matrix solution 10 mg/ml sinapinic acid in 70:30 water/acetonitrile with 0.1% TFA was freshly prepared. 1 µL of the above samples was mixed with 9 µL of the matrix solution, before 1 µL of the final mix was analyzed by MALDI-TOF in biological replicates after air dry. MALDI-TOF spectra of ligand-free 3CL was used as control to determine the mass shift. MALDI spectra were collected with the software flexControl (v3.4) under default settings for protein/peptide analysis (linear positive 5k–50k with a 2 kHz firing rate), while MALDI data were analyzed by flexAnalysis (v3.4).
Mammalian cell-based assay to identify coronavirus protease inhibitor (SARS-CoV, SARS-CoV-2, and MERS)
Expression constructs of SARS-CoV, SARS-CoV-2, or MERS 3CL protease (Twist Biosciences) were cloned into pLEX307 backbone (Addgene #41392) using Gateway LR II Clonase Enzyme mix (Invitrogen, 11791020) and transformed into NEB 10-beta high-efficiency competent cells. DNA plasmids were isolated with miniprep buffers (Omega Biotek, D6942-01) and silica membrane columns (Biobasic, SD5005) before verification by Sanger sequencing (Genewiz). Acquisition of these plasmids was also described previously20.
HEK293T cells were seeded in DMEM supplemented with 10% FBS and PS at approximately 70% confluency into poly-D-lysine-coated 24-well plates, which were coated with 1 mg/mL poly-D-lysine in PBS for 30 min and washed with PBS before seeding. 24 h post seeding, 500 ng of 3CL protease expression plasmids incubated with Opti-MEM and Lipofectamine 2000 for 30 min at room temperature were added to the cells for transfection. 20 h after transfection, cells were collected and homogenized with 200 μL Trypsin for each well. 9 μL of the cell slurry was then seeded into each well of a poly-D-lysine-coated 96-well plate in 100 μL of media and treated with test compound (or DMSO as negative control) and 1 μg/mL puromycin (to select for the transfected cells) for 48 h.
The cells were then washed once with PBS before incubation with 50 μL crystal violet staining solution (0.5% w/v crystal violet in 80% water and 20% methanol) with gentle rocking for 30 min. The cells were then washed four times with water and left to dry for 4 h. 200 μL of methanol was then added for stain extraction via a gentle rocking for 30 min under parafilm seal. 100 μL of the solution was transferred to another 96-well plate for reading 595 nm absorbance in a Tecan Infinite F50 plate reader with the software of Tecan iControl (v.1.10).
Measurement of SARS-CoV-2 viral inhibition in Huh7ACE2 cells
SARS-CoV-2 inhibition was tested as previously described10,36, using a recombinant SARS-CoV-2 (USA-WA1/2020 strain) in which the ORF7a was replaced with a NanoLuc reporter gene37 (gift of Luis Martinez-Sobrido). Huh7ACE2 cells (Huh7 cells overexpressing human ACE2) were produced previously4.
Briefly, 24 h prior to the experiment, 20,000 Huh7ACE2 cells were seeded in 96-well plates in complete media (DMEM + 10% FCS + penicillin/stremptomycin). The next day, test compounds were serially diluted in complete media and added as appropriate to wells in triplicate, along with 0.05 MOI of virus per well. Plates were incubated for a further 24 h, and then cells were lysed and the luciferase activity was quantified using the Nano-Glo® Luciferase Assay System (Promega) according to the manufacturer’s instructions. Inhibition was calculated by comparison to control wells which were uninfected or infected but untreated. EC50 values were then determined by fitting nonlinear regression curves to the data using GraphPad Prism v10.0. All experiments were conducted in a biosafety level 3 (BSL-3) lab.
Measurement of cytotoxicity
24 h prior to test compound treatments, 1000 Huh7ACE2 cells were seeded into each well of a 384-well plate. On the next day, the cells were treated with a two-fold dilution series of test compounds (from 10 µM) and vehicle (DMSO as a negative control) for 48 h. CellTiter-Glo assay reagent (Promega) was added to each well and incubated at room temperature with gentle shaking for 10 min before luminescence was measured in a Tecan Infinite F50 plate reader with the software of Tecan iControl (v.1.10). After normalization to the DMSO control, dose-response curves were analyzed with Prism software (GraphPad, v10.0).
Crystallization, data collection, and structure determination
For crystallization of 3CL protease in complex with inhibitors, we incubated 50 μM 3CL with 500 μM of each inhibitor in the 3CL assay buffer for 1 h at 4 °C, before concentrating it to 5 mg/mL using an ultra-centrifugal filter with 10 kDa molecular weight cutoff (Millipore, UFC5010). With assistance from the High-Throughput Crystallization Screening Center of the Hauptman-Woodward Medical Research Institute (HWI) (https://hwi.buffalo.edu/high-throughput-crystallization-center/), we acquired optimal crystallization conditions for 3CL with DEL7. We reproduced the crystallization condition in house using under oil micro batch method at 4 °C and acquired crystals of 3CL in complex with DEL7. We used these crystals subsequently as seeds for growing crystals of 3CL in complex with all other DEL7 analogs as summarized below.
Block-like crystals of 3CL in complex with inhibitors DEL2, DEL7, 1, 2, 14, 15, 16, 17, 25, 27, 28, 29, 30, 31, 32, and 41 were grown using two crystallization reagents comprising either 0.1 M potassium thiocyanate, 0.1 M sodium acetate (pH 5), and 20% (w/v) PEG 8000 or 0.1 M sodium phosphate monobasic, 0.1 M MES (pH 6), and 20% (w/v) PEG 4000 with protein to crystallization reagent ratio of 2 (or 3):1 µl. All crystals were subsequently transferred into a similar crystallization reagent that was supplemented by 20% (v/v) ethylene glycol or glycerol and flash-frozen in liquid nitrogen. A native dataset was collected on each crystal of the protein-inhibitor complex at two NE-CAT beamlines, 24-ID-C and 24-ID-E, of Advanced Photon Source in Lemont, IL and NSLSII beamlines, AMX, and FMX, of Brookhaven National Laboratory through NECAT BAG users. Crystals of 3CL in complex with inhibitors DEL2, DEL7, 1, 2, 14, 15, 16, 17, 25, 27, 28, 29, 30, 31, 32, and 46 diffracted the X-ray beam to resolution 1.75 Å, 1.69 Å, 2.46 Å, 1.56 Å, 1.55 Å, 1.67 Å, 1.51 Å, 1.72 Å, 1.83 Å, 1.78 Å, 1.71 Å, 1.52 Å, 1.51 Å, 1.52 Å, 1.44 Å, and 1.79 Å, respectively. The images were processed and scaled in space group C2 using XDS (Version February 5, 2021)38. The structure of each protein-inhibitor complex was determined by molecular replacement method using MOLREP (v11.0)39 program and crystal structure of 3CL in complex with GC376 (PDB id: 7JSU) as a search model. After generating a 3D model of each inhibitor using Maestro (Schrödinger Suite, version 2019-1), the LigPrep program of Maestro was used to generate all potential tautomers (isomers) of each inhibitor using default values for OPLS4 force field and ionization states (pH 7.4 ± 2.0). In all cases, Maestro produced several tautomers for each inhibitor. Using program XtalView (v4-1)40, the best plausible hydrogenated tautomer of each inhibitor was then manually docked into the difference (Fo-Fc) electron density map, which was created for each inhibitor after structure determination. Next, Phenix (v1.17.1-3660) elbow was used to generate a CIF file for each inhibitor, followed by editing all bond distances of each inhibitor according to those generated by Maestro. The structure of 3CL with each inhibitor was refined using Phenix41. For covalent inhibitors with an oxygen atom bridging sulfur of C145 and the primary amine of the inhibitors, a covalent 3D model was produced for each inhibitor with C145, followed by manual docking of each inhibitor. All 3CL-inhibitor complexes were crystallized in space group C2 with one protomer and one inhibitor in asymmetric unit of the crystal. The crystallographic statistics are shown in Supplementary Tables 21–24. Given that majority of these structures is at resolution 2 Å or higher, we looked at each inhibitor electron density map at 1-3 σ. If an inhibitor has a complete electron density map throughout its structure at 2 σ, we consider it a strong interactor. Whereas electron density map that is only complete at 1 σ was considered relatively week at this resolution range. The electrostatic surface (KbT/e = ±5.0) of 3CL-inhibitor complex shown in the figures was generated using program APBS42 and rendered in PyMOL (v2.3.4).
Computational docking of 3CL inhibitors for inhibitor optimization
Ligand docking was performed in Maestro (Schrödinger Suite). As a standardized procedure, all compounds were first prepared for in silico docking in the OPLS4 force field by the LigPrep program within Maestro. Possible ionization states at pH 7.0 ± 2.0 and potential tautomers were all generated. The co-crystal structure of 3CL with DEL7 that we determined in this study was used as the receptor for docking. Protein preparation wizard of Maestro was applied to preprocess and refine the protein structure in the OPLS4 force field, before generation of the receptor grid. The DEL7 ligand in the co-crystal structure was used to define the centroid of docking site before being excluded from the grid generation. Ligand docking with extra precision was performed by the Glide program within Maestro. The docking was constrained to restrict docking to a reference position defined by DEL7 in the binding site of the co-crystal structure with 0.1 Å tolerance and intramolecular hydrogen bonds rewarded. Top docking score for each compound is reported, for which a lower value indicates a higher binding affinity. For example, a compound with a docking score of −8 is predicted to bind to 3CL with higher affinity than a compound with a score of −5. The scores of compounds were used to guide the medicinal chemistry optimization of DEL7.
Surface plasmon resonance (SPR) binding assay
The binding affinity of inhibitors for 3CL was assayed using the SPR-based Biacore X100 instrument (Cytiva). His-tagged 3CL (3CL-GP-6xHis in which GP is a linker between 3CL and His-tag) was immobilized on a CM5 sensor chip by using His Capture Kit (Cytiva, #28995056) under standard condition at 25 °C with running buffer HBS-EP+ (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.05% v/v surfactant P-20). A reference flow cell was activated and blocked in the absence of 3CL. The 3CL immobilization level was fixed at 1000 response units (RU), and then different concentrations of inhibitors were serially injected into the channel to evaluate binding affinity. Between injections of inhibitors of different concentrations, regeneration of sensor chip was performed by washing with the regeneration buffer (20 mM glycine, pH 1.5) provided by the same His Capture Kit (Cytiva, #28995056). The equilibrium dissociation constant (KD) of the inhibitors was obtained by fitting binding response units to the Hill equation.
Microsome stability assays
1 µM test compounds or control compounds was incubated with 0.5 mg/ml liver microsomes (human or CD-1 mouse) in PBS supplemented with an NADPH regenerating system (Promega) at 37 °C under gentle rotation. 10 µL of the mixture was added into 90 µL ice-cold acetonitrile that contains an internal standard. We quantified the level of test compounds or control compounds in the samples using the software of MassLynx (v4.2) to set up a gradient of 23–100% B (A: water + 0.2% formic acid, B: 4:1 methanol/isopropanol) in ESI+ mode on a LC-MS equipped with a Waters H-Class Plus UPLC (Acquity UPLC BEH C18, 2.1 × 50 mm, 1.7 µm) connected to a Xevo G2-XS QTOF mass spectrometer.
Cell permeability assay
For each 3CL inhibitor, HEK-293T cells were seeded at 1 million cells per well into 6 wells of a 6-well plate and incubated overnight. The cells were then treated with 10 µM 3CL inhibitors on the next day. To measure whole-cell average concentration of the 3CL inhibitor, after 4 h incubation with the inhibitor, 3 wells of cells were separately harvested with trypsin (Invitrogen, 25200-114), pelleted, washed with PBS, and counted on a Vi-Cell XR Cell Viability Analyzer (Beckman Coulter). The average diameter of cells was also recorded by the analyzer. Number of cells and the average diameter were used to calculate the total volume of cells from each volume, assuming that cells are spherical in suspension state. Half of the cells from each well were pelleted separately at 3000 rpm for 3 min, after which the PBS was removed and the pellet frozen at −80 °C overnight. The cell pellets were defrosted on the next day and 500 µL of methanol was added to each pellet. The suspension was sonicated for 10 s and the compound was extracted overnight at 4 °C. The samples were spun at 1400 g for 75 min at 4 °C, and the supernatant (250 µL) was withdrawn and transferred to 3 sample vials with insert. The samples were analyzed by LC-MS to determine whole-cell concentration of the compound.
To measure the cytosolic concentration of 3CL inhibitors, after 4 h incubation with the inhibitor, cell culture media were removed from the other 3 wells and then 1 mL solution of 20 µg/mL digitonin in PBS was added to each of the 3 wells to permeabilize the cell membrane to release cytosol fraction but not organelles nor nuclear. It is noteworthy that 1 mL of solution (digitonin) was used here on each well while 0.5 mL of solution (methanol) was used above on half cells from each well to enable a side-by-side comparison. The plate was kept at room temperature (25 °C) for 5 min before shaking on ice for 30 min on an orbital shaker. 500 µL of solution from each well and analyzed by LC-MS to determine cytosolic concentration of the compound. UPLC-MS analysis was performed on Synapt G2-XS (Waters Corp., U.K) mass spectrometer, equipped with an electrospray ionization source. Chromatographic separation was performed by injecting 1 µL of the sample onto an Acquity Premier BEH C18 column (1.7 µm, 2.1 × 50 mm) maintained at 50 °C, over a 3 min gradient. The flow rate was maintained at 0.5 mL/min. The initial flow conditions were 50% solvent A (water containing 0.1% formic acid) and 50% solvent B (methanol containing 0.1% formic acid) and the gradient was held for 0.25 min. Solvent B was raised to 100% by 1 min and returned to the initial condition by 2.00 min, and the column was equilibrated for additional 1.00 min. The data were acquired in positive ionization mode, over a mass range of m/z 50–1000, using the following parameters—capillary voltage of 0.5 kV, sampling cone voltage of 32 V, source temperature of 120 V, desolvation temperature of 400 °C and desolvation gas flow rate of 800 L/h. Leucine enkephalin (m/z 556.2771 [M + H]+) was used as a lock mass via lockspray interface for mass accuracy. The compound concentration in each sample was determined using QuantLynx of MassLynx (Waters, v4.2) with a standard calibration curve of each compound. In particular, the base peak chromatogram at m/z 598.17 (corresponding to [M + H]+ of DEL7), 683.26 (corresponding to [M + H]+ of 7), or 622.17 (corresponding to [M + H]+ of 18) with a width of ±0.02 was integrated and peak area was quantified.
Measurement of HCoV-Alpha-229E, HCoV-Beta-OC43, and SARS-CoV-2 (Omicron variant) viral inhibition
Huh7 (for HCoV-229E), RD (for HCoV-OC43), and Caco-2 (for Omicron variant of SARS-CoV-2) cells were seeded in Minimum Essential Medium (MEM) supplemented with 2% FBS and 50-µg/ml gentamicin at approximately 90% confluency into 96-well plates. 24 h post seeding, the test compound or DMSO (as vehicle negative control) was added at a serial diluted concentrations in triplicates. EIDD-1931 was used as a positive drug control in addition to an untreated and uninfected positive control. 0.003 MOI of HCoV-Alpha-229E or HCoV-Beta-OC43, or SARS-CoV-2 (XBB lineage, hCov-19/USA/CA-Stanford-109_S21/22 strain) at 50 CCID50 (50% cell culture infectious dose) was also added to the cells. When >80% CPE (virus-induced CytoPathic Effect) was observed in infected cells treated with DMSO, all cells were stained with 0.011% neutral red for 2 h at 37 °C before a wash with PBS. 50% Sorensen’s citrate buffer and 50% ethanol was added for an incubation of 30 min before reading 540 nm absorbance in a Tecan Infinite F50 plate reader with the software of Tecan iControl (v.1.10).
Statistical information
The data reported in this study represent the mean and s.d. of at least three independent experiments measured in triplicate, unless otherwise stated in the figure legend.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Structural data for the SARS-CoV-2 3CL protease in complex with DEL2, DEL7, 1, 2, 14, 15, 16, 17, 25, 27, 28, 29, 30, 31, 32, and 46 were deposited in the Protein Data Bank (PDB), with accession codes PDB IDs: 8UDJ, 8UDF, 8UDQ, 8UDW, 8UDP, 8UDO, 8UDM, 8UDX, 8UDY, 8UEG, 8UEI, 8UEA, 8UEB, 8UEH, 8UEF, and 8UE0. Publicly available dataset used in this study is the crystal structure of SARS-CoV-2 3CL protease without any ligand or in complex with GC376, compound 4, nirmatrelvir, S-217622, WU-04, GC-14, JZD-07, Compound 23 R, and ML188, with accession code PDB IDs: 7JST, 7JSU, 7JT7, 7U28, 7VU6, 7EN8, 8ACL, 8GTV, 7KX5, and 7L0D. All other data generated in this study are provided in the Supplementary Information, and are available upon request from the corresponding author. Source data are provided with this paper.
References
Kupferschmidt, K. & Wadman, M. End of COVID-19 emergencies sparks debate. Science 380, 566–567 (2023).
WHO. WHO Coronavirus (COVID-19) Dashboard, https://covid19.who.int (2023).
Lau, J. J. et al. Real-world COVID-19 vaccine effectiveness against the Omicron BA.2 variant in a SARS-CoV-2 infection-naive population. Nat. Med. 29, 348–357 (2023).
Liu, H. et al. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nat. Commun. 13, 1891 (2022).
Jin, Z. et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 368, 409–412 (2020).
Owen Dafydd, R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Hammond, J. et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N. Engl. J. Med. 386, 1397–1408 (2022).
Unoh, Y. et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 65, 6499–6512 (2022).
Iketani, S. et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 613, 558–564 (2023).
Zuckerman, N. S., Bucris, E., Keidar-Friedman, D., Amsalem, M. & Brosh-Nissimov, T. Nirmatrelvir resistance-de novo E166V/L50V mutations in an immunocompromised patient treated with prolonged nirmatrelvir/ritonavir monotherapy leading to clinical and virological treatment failure-a Case Report. Clin. Infect. Dis. 78, 352–355 (2024).
Hirotsu, Y. et al. Multidrug-resistant mutations to antiviral and antibody therapy in an immunocompromised patient infected with SARS-CoV-2. Med. 4, 813–824.e4 (2023).
Iketani, S. et al. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun. 12, 2016 (2021).
Yang, S. et al. Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med Chem. 49, 4971–4980 (2006).
Valley, C. C. et al. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J. Biol. Chem. 287, 34979–34991 (2012).
Rabe von Pappenheim, F. et al. Widespread occurrence of covalent lysine–cysteine redox switches in proteins. Nat. Chem. Biol. 18, 368–375 (2022).
Steuten, K. et al. Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID−19. ACS Infect. Dis. 7, 1457–1468 (2021).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 e278 (2020).
Zhao, M.-M. et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 6, 134 (2021).
Resnick, S. J. et al. Inhibitors of coronavirus 3CL proteases protect cells from protease-mediated cytotoxicity. J. Virol. 95, e0237420 (2021).
Labunskyy, V. M., Hatfield, D. L. & Gladyshev, V. N. Selenoproteins: molecular pathways and physiological roles. Physiol. Rev. 94, 739–777 (2014).
Hou, N. et al. Development of highly potent noncovalent inhibitors of SARS-CoV-2 3CLpro. ACS Cent. Sci. 9, 217–227 (2023).
Han, S. H. et al. Structure-based optimization of ML300-derived, noncovalent inhibitors targeting the severe acute respiratory syndrome coronavirus 3CL protease (SARS-CoV-2 3CL(pro).J Med Chem 65, 2880–2904 (2022).
Gao, S. et al. Discovery and crystallographic studies of trisubstituted piperazine derivatives as non-covalent SARS-CoV-2 main protease inhibitors with high target specificity and low toxicity. J. Med Chem. 65, 13343–13364 (2022).
Jiang, Z. et al. Discovery of novel non-peptidic and non-covalent small-molecule 3CL(pro) inhibitors as potential candidate for COVID-19 treatment. Signal Transduct. Target Ther. 8, 209 (2023).
Herlah, B. et al. Design, synthesis and evaluation of fused bicyclo[2.2.2]octene as a potential core scaffold for the non-covalent inhibitors of SARS-CoV-2 3CL(pro) Main Protease. Pharmaceuticals 15, 539 (2022).
Ma, C. et al. Discovery of Di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 143, 20697–20709 (2021).
Lockbaum, G. J. et al. Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses 13, 174 (2021).
Lee, J. Y. et al. Identification of non-covalent 3C-like protease inhibitors against severe acute respiratory syndrome coronavirus-2 via virtual screening of a Korean compound library. Bioorg. Med. Chem. Lett. 42, 128067 (2021).
Ferreira, J. C., Fadl, S. & Rabeh, W. M. Key dimer interface residues impact the catalytic activity of 3CLpro, the main protease of SARS-CoV-2. J. Biol. Chem. 298, 102023 (2022).
Chen, H. et al. Only one protomer is active in the dimer of SARS 3C-like proteinase. J. Biol. Chem. 281, 13894–13898 (2006).
Goyal, B. & Goyal, D. Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 22, 297–305 (2020).
Satz, A. L. et al. DNA-encoded chemical libraries. Nat. Rev. Methods Prim. 2, 3 (2022).
Zhu, Z. et al. Development of a selection method for discovering irreversible (covalent) binders from a DNA-encoded library. SLAS Discov. 24, 169–174 (2019).
Shimada, K. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016).
Iketani, S. et al. Functional map of SARS-CoV-2 3CL protease reveals tolerant and immutable sites. Cell Host Microbe 30, 1354–1362.e1356 (2022).
Ye, C. et al. Rescue of SARS-CoV-2 from a single bacterial artificial chromosome. mBio 11, https://doi.org/10.1128/mBio.02168-20 (2020).
Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D. Biol. Crystallogr 66, 133–144 (2010).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr D. Biol. Crystallogr 66, 22–25 (2010).
McRee, D. E. XtalView/Xfit-A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125, 156–165 (1999).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr 66, 213–221 (2010).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl Acad. Sci. 98, 10037–10041 (2001).
Acknowledgements
This work was supported by grants from the Jack Ma Foundation and NIAID (1U19AI1711401) to D.D.H. and A.C. and by grants from Columbia Technology Ventures and the Columbia Translational Therapeutics (TRx) program to B.R.S. A.C. is also supported by a Career Awards for Medical Scientists from the Burroughs Wellcome Fund. S.I. was supported by NIH grant T32AI106711. E.B. thanks the Experientia Foundation for a postdoctoral fellowship. We thank the staff of the High-Throughput Crystallization Screening Center of the Hauptman-Woodward Medical Research Institute for screening of crystallization conditions. Crystallization screening was supported through NSF grant 2029943. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The Center for Bio-Molecular Structure (CBMS) is primarily supported by the NIH-NIGMS through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1607011). NSLS2 is a U.S.DOE Office of Science User Facility operated under Contract No. DE-SC0012704. This publication resulted from the data collected using the beamtime obtained through NECAT BAG proposal # 311950. Extraordinary facility operations were supported in part by the DOE Office of Science through the National Virtual Biotechnology Laboratory, a consortium of DOE national laboratories focused on the response to COVID-19, with funding provided by the Coronavirus CARES Act. Funding for experiments completed at Utah State University was provided by the Respiratory Diseases Branch, National Institute for Allergy and Infectious Diseases, NIH USA (Contract N01-AI-30048).
Author information
Authors and Affiliations
Contributions
B.R.S. conceived and implemented the project. H.L., A.Z., F.F., and B.R.S. planned and designed the experiments. S.I. and S.J.H. cloned, expressed, and purified proteins. H.L. conducted the DEL library screen. H.L. conducted the protease inhibition assay. S.I., D.H.L, T.B., and D.D.H. devised and performed the Huh7ACE2-based antiviral assay. H.L. performed the human protease selectivity assay. H.L performed mass spec analysis of protein-inhibitor complex and cell permeability. S.J.R. and A.C. devised and performed the HEK293T-based protease inhibitor assay. H.L. and F.F. crystallized the proteins. F.F. collected diffraction data and solved the crystal structures. F.F. and H.L. analyzed the structural data. A.Z., F.F., H.L., A.W., D.R.V., D.L.H., and K.C. designed and synthesized analogs of DEL7. H.L., F.F., A.Z., and B.R.S. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
S.I., H.L., A.Z., F.F., B.R.S., A.C., and D.D.H. are inventors on a patent application submitted based on this work. B.R.S. is an inventor on additional patents and patent applications related to small molecule therapeutics, and co-founded and serves as a consultant to Exarta Therapeutics, and ProJenX Inc., serves as a consultant to Weatherwax Biotechnologies and Akin Gump Strauss Hauer & Feld LLP, and holds equity in Sonata Therapeutics. S.I. and D.D.H. have sponsored research agreements with Enanta Pharmaceuticals, Shionogi & Co., Ltd., and Regeneron Pharmaceuticals. D.D.H. is a co-founder of TaiMed Biologics and RenBio, consultant to WuXi Biologics, Brii Biosciences, Apexigen, and Veru Inc., and board director for Vicarious Surgical. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, H., Zask, A., Forouhar, F. et al. Development of small molecule non-covalent coronavirus 3CL protease inhibitors from DNA-encoded chemical library screening. Nat Commun 16, 152 (2025). https://doi.org/10.1038/s41467-024-55421-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55421-5
