
Abstract
Since SARS-CoV-2’s emergence, studies in Southeast Asia, including Cambodia, have identified related coronaviruses (CoVs) in rhinolophid bats. This pilot study investigates the prevalence and diversity of CoVs in wildlife from two Cambodian provinces known for wildlife trade and environmental changes, factors favoring zoonotic spillover risk. Samples were collected from 2020 to 2022 using active (capture and swabbing of bats and rodents) and non-invasive (collection of feces from bat caves and wildlife habitats) methods. RNA was screened for CoVs using conventional pan-CoVs and real-time Sarbecovirus-specific PCR systems. Positive samples were sequenced and phylogenetic analysis was performed on the partial RdRp gene. A total of 2608 samples were collected: 867 rectal swabs from bats, 159 from rodents, 41 from other wild animals, and 1541 fecal samples. The overall prevalence of CoVs was 2.0%, with a 3.3% positive rate in bats, 2.5% in rodents, and no CoVs detected in other wildlife species. Alpha-CoVs were exclusive to bats, while Beta-CoVs were found in both bats and rodents. Seven SARS-CoV-2-related viruses were identified in Rhinolophus shameli bats sampled in August 2020, March 2021, and December 2021. Our results highlight diverse CoVs in Cambodian bats and rodents and emphasize bats as significant reservoirs. They also suggest continuous circulation of bat SARS-CoV-2-related viruses may occur in a region where ecological and human factors could favor virus emergence. Continuous surveillance and integrated approaches are crucial to managing and mitigating emerging zoonotic diseases.
Similar content being viewed by others
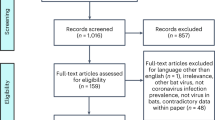

Introduction
SARS-CoV-2, the virus responsible for the COVID-19 pandemic, first emerged in late 2019 in the city of Wuhan, Hubei Province, China1. The virus belongs to the family Coronaviridae and its closest known relatives are found in bats, particularly horseshoe bats (Rhinolophus spp.)1,2. Over the past two decades, it is the third coronavirus originating from animals to infect humans and led to a pandemic that has had profound and far-reaching impacts on global health, economies, and societal well-being. Recognition of wildlife as reservoirs for coronaviruses (CoVs) has grown significantly in recent decades3 and bats in particular have been identified as carriers of a wide variety of CoVs, including those related to SARS-CoV, MERS-CoV, and SARS-CoV-21,4,5,6. CoVs of veterinary importance have also been traced back to bats, such as SADS-CoV, an important pig pathogen, for which a 98% identity was found with HKU2 bat CoV7. Rodents have also been implicated as reservoirs for certain CoVs, including Beta-CoVs causing mild illness in humans such as the now endemic HKU13. Many mammal and avian species are susceptible to a wide range of CoVs and Beta-CoVs have infected free-ranging and captive individuals of wild animals including rodents, civets, minks, ferrets, deer and non-human primates, as well as domestic animals such as cattle, dogs and cats8. Most of the human-endemic CoVs have a reported intermediate species3,9,10. Phylogenetic evidence suggests that both hCoV-OC43 and hCoV-HKU1 might have originated from rodents, with cattle and camelids proposed as intermediate hosts9,11. On the other hand, hCoV-NL63 and hCoV-229E are thought to have a bat origin, with no evidence of intermediate hosts. Pathogenic human CoVs such as SARS-CoV and MERS-CoV are believed to have been transmitted to humans through intermediate amplifying hosts, specifically masked palm civet and raccoon dogs for SARS-CoV and dromedary camels for MERS-CoV3,10. While the animal origins of SARS-CoV-2 are still debated, SARS-CoV-2 related viruses have been found in various wild species including horseshoe bats, pangolins and racoon dogs12.
From a global perspective, the complex interactions between anthropogenic environmental changes and other human activities such as legal or illegal wildlife trade have created unique challenges to understanding and managing risks associated with pathogen emergences, especially for CoVs13,14,15. In Southeast Asia particularly, substantial environmental modifications have occurred16 and several human activities can facilitate the spillover of viruses, such as guano farming, intensive livestock farming, agriculture expansion15,17, deforestation, and wildlife farming and breeding, trading of wildlife species or activities such as hunting and butchering18,19,20. In Vietnam for instance, Huong et al. highlighted a high prevalence of CoV and amplification process along the supply chain for rodent consumption21. Similarly, Cantlay et al. found a high diversity of potentially zoonotic pathogens in wild animal species hunted for consumption in Malaysia, especially among members of the Cercopithecidae, Suidae and Cervidae18.
Since the emergence of SARS-CoV-2, significant efforts have been directed towards identifying CoVs reservoirs and finding the possible ancestor of SARS-CoV-2. In identifying animal reservoirs and assessing their extent, crucial steps can be taken to better understand and ultimately control the risks of emergence of zoonotic pathogens. Studies have revealed significant geographical variation in the prevalence and types of CoV found especially in bat populations and strains related to SARS-CoV-2 have been identified in the Indochinese Peninsula of Southeast Asia, China, Laos, Thailand and Cambodia22,23,24,25,26. Further, a virus found in Cambodia in two Rhinolophus shameli bats sampled in northern Cambodia in 2010 shared a 92.6% nucleotide identity with SARS-CoV-222. As such, the escalation in human-driven changes that Cambodia has experienced in recent years, such as extensive deforestation and agricultural expansion and rampant trade and consumption of rats and other wildlife species, could pose a substantial risk of zoonotic spillover events21,27,28,29.
A One-Health transdisciplinary project (ZooCoV) was initiated in 2020 in Cambodia to work towards integrated surveillance of potentially zoonotic CoVs in wild animal value chains in Cambodia30. To investigate the complex landscape of CoVs in Cambodia, samples were collected from various wildlife species in Stung Treng and Mondulkiri, two provinces experiencing significant wildlife trade and land-use changes31,32,33. Additionally, two bat CoVs related to SARS-CoV-2 have been detected in horseshoe bats samples collected in 2010 in Stung Treng area, where karst hills are hosting large colonies of Rhinolophus spp., believed to be the main reservoir hosts of SARS-related coronavirus - Sarbecovirus, a subgenus of Beta-CoVs22,34. We present here the results of an exploratory study which aimed to characterize the prevalence and diversity of CoVs in the interfaces between human and wild animal populations in two provinces of Cambodia.
Methods
Study sites
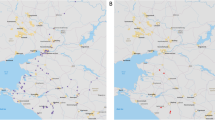
The Mondulkiri and Stung Treng provinces were selected for our study because trade and consumption of wildlife, extensive human-driven environmental changes35, and occurrence of bat colonies hosting viruses related to SARS-CoV-2 have been documented in these areas22,34. Mondulkiri province is located in eastern Cambodia, and is the largest province in the country at 14,288 km2 (Fig. 1). The province has several protected areas, including Keo Seima Wildlife Sanctuary which has experienced extensive deforestation and resource extraction36. Stung Treng province is located in northeast Cambodia, on the international border with Laos. The province is largely forested, although timber logging, fishing, and agricultural encroachment have placed strain on the natural resources of the region37. Several karst hills exist in the province which host cave bat colonies of various species, including horseshoe bats22,34. In each province, several sites were chosen for active and/or non-invasive sampling sessions (Fig. 1). In this context, we define active sampling as the capture of wild animals or the handling of captive wild species and their sampling, whereas non-invasive sampling comprises the collection of animal feces from the environment.

Location of Mondulkiri and Stung Treng provinces in Cambodia and the location and timing of sampling efforts for study species.
Ethics approval
All applicable institutional and/or national guidelines for the care and use of animals were followed and only trained personnel undertook procedures of handling and sampling the animals. All animals were captured and handled in accordance with the Guidelines of the American Society of Mammologists for the use of wild mammals in research and education38 and the American Veterinary Medical Association Guidelines for the Euthanasia of Animals39, in addition to the requirements of the statutory study permission provided by the national authority responsible for wildlife research, i.e.: the Forestry Administration (FA) of the Cambodian Ministry of Agriculture, Forest and Fisheries. A study agreement was obtained from the FA who participated in and oversaw all aspects of active sampling during the field investigations, because an animal ethics committee did not exist in Cambodia. This study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org/ )40.
Active sampling
Bat trapping and sampling
Bats were live-captured around four karst hills in the Thala Borivat District of Stung Treng province, in areas described by Delaune et al.22. Each hill was investigated on two non-consecutive nights during five sampling sessions undertaken in August and October 2020 and March, September and December 2021 (Fig. 1). Bats were captured before their emergence at dusk until 8:00 pm each night using mist-nets deployed at the edges of forests and entrances of caves and in surrounding areas. These were immediately placed in individual cloth bags for subsequent processing. All bats captured were examined to determine their specific identity (based on morphological criteria) and their sex, age and reproductive status were registered41,42. Two rectal swabs were collected from each animal before its release at the capture site. Swabs were individually stored in TRIzol™ reagent (Sigma-Aldrich) and in-house viral transport medium (VTM, composition described in supplemental file 1). All samples were stored in liquid nitrogen post-collection, then at -80 °C at the Virology Unit of Institut Pasteur du Cambodge (IPC) until subsequent analysis.
Rodent trapping and sampling
Rodents were live-trapped in February 2021 (Mondulkiri province) and March 2021 (Stung Treng province) in selected locations (Fig. 1) where ongoing serological human surveys have been undertaken as part of the broader ZooCoV project (the latter will be reported elsewhere)30. Sixty locally-made live traps were deployed over one to two consecutive nights. In each province, the main road of a village was chosen and house-to-house visits were undertaken with the support of local authorities to deploy the traps within and outside of individual households in the evening. These were baited with a mixture of steamed corn, sausages, peanut butter, dried fish and sweet potatoes. Additional trapping sessions were undertaken inside and around three bat caves over one night in Stung Treng in March, September and December 2021 using 20 live-traps (Fig. 1). In each instance, a sampling station was established in the morning and rodents were humanely euthanized by trained personal in open-air settings using an overdose of inhalant anesthetic in accordance with the Guidelines of the American Society of Mammologists for the use of wild mammals in research and education38. Briefly, each animal was transferred into a non-rigid container containing cotton ball soaked with isoflurane (0.4mL for 20 g estimated body mass). Death was confirmed by ascertaining cardiac and respiratory arrest and the observation of fixed and dilated pupils. Following euthanasia, organs were dissected, and oral and rectal swabs were collected and individually stored in VTM following Herbreteau et al.43. Samples were stored in liquid nitrogen post-collection, then at -80 °C at the Virology Unit of IPC until subsequent analysis.
Opportunistic wildlife sampling
Collaboration with non-government organizations (NGOs) and private institutions allowed the collection of additional samples from various animal species potentially in contact with general public in both provinces (Fig. 1): (i) captive and/or semi-captive wild animals in Mondulkiri province with the support of both the Elephant Valley Project44 and the Mayura Wild Park (GPS coordinates: 12.62924 N, 107.25591 E), (ii) animals found dead or trapped in Stung Treng province45. Only rectal/cloacal swabs or fresh feces were collected and individually stored in VTM as previously described. Further information on collaborating organizations and the collection of these samples is provided in supplemental file 2.
Non-invasive sampling
Bat feces collection
Concurrent to the live-capture of bats and sampling sessions in Stung Treng province, bat feces were collected from the floor of caves where trapping was undertaken (Fig. 1). Individual caves supported either insectivorous bats only, frugivorous bats only or both. Visually fresh feces were collected and placed individually in cryotubes containing VTM. All samples were stored in an icebox then in liquid nitrogen in the field for 24 h post-collection, then at -80 °C at the Virology Unit at the IPC until analysis.
Wildlife feces collection
We collaborated with the Jahoo NGO46 to collect fresh feces from wildlife, including bats, within the Keo Seima Wildlife Sanctuary from March 2021 to February 2022. Jahoo rangers were provided with individual cryotubes containing 1.0 mL of RNAlater™ stabilization solution (Qiagen) and trained for safe collection of fresh feces. Pea-sized individual feces were collected and information on the location and estimated taxa/species were indicated. A focus was requested on specific orders —Chiroptera, Primates, Carnivora and Artiodactyla— but all other feces encountered were also collected. Samples were stored at -20 °C on-site and sent monthly to the Virology Unit at the IPC, where they were stored at -80 °C until further analysis.
Molecular detection of coronavirus
Total RNA of rectal swab samples was extracted using Zymo Research Direct-zol RNA MiniPrep kit (Zymo Research, Cat # R2050, California, USA) according to the manufacturer’s instructions. Bat feces collected in Stung Treng Province were homogenized and pooled by five feces prior to extraction. No pooling was performed for feces collected from wildlife. Following cDNA synthesis using SuperScript III First-Strand Synthesis Super-Mix (Invitrogen, California) according to the manufacturer’s instructions, two pan-Coronaviridae conventional RT-PCR targeting the RdRp gene was used for screening as previously described41,47,48. RNA samples were additionally screened using an adapted duplex one-step real-time PCR targeting E and N genes of Sarbecovirus (Supplemental file 3)49. Whenever a pool of bat feces was positive for CoV detection with one of the detection systems, testing of individual fecal sample from that pool was performed. All PCR amplification-positive samples were subsequently confirmed by Sanger sequencing (Macrogen, Inc., Seoul, Republic of Korea) in both forward and reverse directions using the primers from the second round of nested PCR for conventional molecular assays. The sequences obtained were confirmed by similarity using the NCBI BLASTn search (https://www.ncbi.nlm.nih.gov/BLAST). A sample was considered positive for CoV if one of the PCR systems presented confirmed positive result.
Additionally, positive samples were submitted to DNA barcoding to confirm species of origin using conventional PCR protocols targeting vertebrate mitochondrial cytochrome oxidase subunit 1 (CO1) and cytochrome b (Cyt b) mtDNA as described by Towzen et al.50. Sanger sequencing was performed on PCR products (Macrogen Inc., Seoul, Republic of Korea) for both forward and reverse strands. The specific identity of each specimen was determined by comparing each sequence with homologous sequences contained in GenBank (BLASTn tool).
Phylogenetic analysis
Two datasets were used to perform the phylogenetic analyses based on the region sequences obtained with Quan et al. and/or Watanabe et al. protocols. In both cases, sequences were aligned using online Multiple alignment program for amino acid or nucleotide sequences (MAFFT Version 7, https://mafft.cbrc.jp/alignment/server/index.html)51 with a subset of selected reference sequences (RefSeq) available in Coronavirus GenBank database (https://www.ncbi.nlm.nih.gov/datasets/coronavirus/genomes/). Consequently, the “Quan dataset” corresponds to a 295-nt sequence alignment including 43 sequences from this study and 65 reference sequences, whereas the “Watanabe dataset” corresponds to a 389-nt sequence alignment including 15 sequences from this study and 117 reference sequences. Phylogenetic trees using the two datasets were inferred using the maximum-likelihood method in IQ-TREE v2.0.652, and branch support was calculated using the ultrafast bootstrap approximation with 1000 replicates53. Prior to tree reconstruction, the ModelFinder application within IQ-TREE was used to select the best-fitting nucleotide substitution model54.
Results
Study samples characteristics
In total, 2,608 samples were collected between October 2020 and February 2022 (Table 1). Our active sample comprised 1,067 individual rectal swabs from 867 (81.3%) bats, 159 (14.9%) rodents and 41 (3.8%) other wild animals, representing 40.9% of the total study sample. Bats belonging to five families and eight genera were captured, including Rhinolophus (65.5%, 568/867 individuals), Taphozous (20.2%, 175), Hipposideros (5.4%, 47), Cynopterus (3.1%, 27), Rousettus (2.0%, 17), Megaderma (1.7%, 15), Lyroderma (1.6%, 14) and Eonycteris (0.5%, 4). A total of 159 rodents were sampled, with Rattus exulans the most frequently captured species (58.5%, 93/159). This was followed by an unidentified Rattus sp. (39.0%, 61/159) and three Rattus norvegicus (1.9%). Additionally, one individual of Vandeleuria oleracea was sampled from a cave in Stung Treng. All R. exulans were captured in or around households, primarily in villages sampled in Stung Treng (47.3%, 44/93) and Mondulkiri (52.7%, 49/93) provinces. The Rattus sp. individuals were exclusively captured within or near bat caves in Stung Treng, whereas R. norvegicus were found solely in or around households in the same province. Other wild animals opportunistically sampled included Elephantidae (n = 7), Cervidae (n = 5), Suidae (n = 26), Cercopithecidae (n = 1) and Accipitridae (n = 2).
Our non-invasive sample comprised 1,541 individual feces or 59.1% of the total study sample. Bat feces accounted for 87.0% (1,341/1,541) of these. Among those bat feces, 1,170 feces (87.2%) were collected inside karst caves in Stung Treng province and 171 bat feces (12.8%) were collected under tree roosts in Mondulkiri province. Of other wildlife feces collected (n = 200), most were from Primates (n = 111), Carnivora (n = 63) and Rodentia (n = 9) (supplemental file 4).
CoV prevalence and diversity
CoV prevalence
The overall prevalence of CoVs in our study samples was 2.0% (51/2608, CI-95 1.4–2.5) using both conventional and real-time PCR protocols (Table 1). Of the 51 positive samples, 35 were detected with the “Quan” protocol only and three with the “Watanabe” protocol only. Eight samples were detected simultaneously by both conventional PCR systems. Of the 9 samples positive with the real-time PCR system, all were detected positive using the “Watanabe” protocol while only four were detected by the “Quan” protocol. CoV were only detected in samples from bats and rodents accounting for a total prevalence of 3.1% (33/1067, CI95 2.1–4.1) among active collection samples, and 1.2% (18/1541, CI95 0.6–1.7) among non-invasive collection samples.
Among live-captured bats sampled in Stung Treng province, CoV prevalence was 3.3% (29/867, CI95 2.1–4.5) and CoVs were detected at every active session of sampling (Supplemental file 5). Apart from the Pteropodidae, all bat families were positive for at least one CoV: 8.5% (4/47) among Hipposideridae, 3.9% (22/568) among Rhinolophidae, 3.4% (1/29) among Megadermatidae, and 0.6% (1/175) among Emballonuridae. Among Rhinolophus bats, CoVs were detected in R. microglobosus and R. shameli, with prevalence of 42.9% (6/14, CI95 16.9–68.8) and 3.4% (17/505, CI95 1.8–4.9), respectively. H. larvatus s.l. and H. gentilis were the two species where CoVs were detected among Hipposideridae family, with respective prevalence of 13.0% (3/23, CI95 0.0-26.8), and 33.3% (1/3, CI95 0.0-86.7).
Overall prevalence was 2.5% (4/159, CI95 0.1–4.9) among rodents and CoVs were solely detected in the Rattus genus, including 3.2% (3/93, CI95 0.0-6.8) in R. exulans trapped in and around households in Mondulkiri and 1.6% (1/62, CI95 0.0-4.7) among one Rattus sp. trapped inside one of the bats caves in Stung Treng.
CoVs were detected among environmental bat feces collected in Stung Treng province only, and none of wildlife feces from Mondulkiri province was positive. Prevalence of CoV among bats feces was 1.3% (17/1,341, CI95 0.7–1.9). Among feces collected from additional wildlife species, one CoV was detected (1/199, CI95 0.0-1.5) in a rodent’s feces (Mus cervicolor) collected in one of the bat caves in Stung Treng province.
CoV diversity
In total, 43 sequences obtained using “Quan” PCR system and 13 sequences obtained using “Watanabe” protocol were used to perform phylogenetic analysis (Fig. 2A, B). Because three of them presented with ambiguous nucleotides sequences, they were removed from the analysis. The GenBank accession numbers of CoVs detected in this study and included in our phylogenetic analyses are available in supplemental file 6. Based on phylogenetic analysis of partial RdRp gene, CoVs detected in this study belong to Alpha-CoV (n = 21) and Beta-CoV (n = 29) genera. All Alpha-CoV were detected from bat rectal swabs except one from feces. They were classified into three distinct phylogenetic groups: Minunacovirus, Decacovirus and Rhinacovirus. The two viral groups belonging to Minunacovirus and Decacovirus were related to BatCoV strains AFCD62 (Genbank: EU420138) and HKU10 (Genbank: NC_018871), respectively, in the “Quan” dataset, despite substantial divergence among sequences. Additionally, the viral group belonging to Rhinacovirus was highly similar to BatCoV HKU2/4646 (Genbank: EF203065). One sequence of CoV detected in a R. shameli rectal swab clustered within the Minunacovirus lineage, along with sequences obtained from H. larvatus. Sequences obtained with “Watanabe” protocol originated from rectal swabs collected from R. shameli individuals. Two available sequences belonged to Decacovirus (HKU10-related) supported by a 99% bootstrap value, while two others were related to Rhinacovirus BatCoV-HKU2, with 100% bootstrap support. Beta-CoVs were detected in rectal swabs from bats and rodents, as well as bat feces, and were classified into the Embecovirus (n = 5), Sarbecovirus (n = 8), and Nobecovirus (n = 13) subgenera. Eight viruses from bat’s rectal swabs within Sarbecovirus were found in the SARS-CoV-2 sub-lineage and were closely related to RshSTT182 and RshSTT200, detected in the same area in 2010 in R. shameli species22. Four of them were detected using the “Quan” system while all of them were detected using the “Watanabe” system. Six of these viruses were detected from rectal swabs of horseshoe bats (R. shameli): one in August 2020 from a mature male, one in March 2021 from an immature male, and four in December 2021 comprising one parous female and three mature males. Two others were detected from rectal swabs of black-bearded tomb bats (Taphozous melanopogon, parous female) and lesser false vampire bats (Megaderma spasma, nulliparous female) sampled in December 2021. All Beta-CoVs of Embecovirus subgenus were detected using the “Quan” system in samples from rodents and were related to RatCoV Parker (GenBank: FJ938068) (n = 4) and RatCoV Longquan-343-Aa (GenBank: KF294357) (n = 1). Beta-CoVs of Nobecovirus subgenus were detected only in bat feces from both Rousettus spp. and insectivorous bats (T. melanopogon and other unidentified) and were closely related to BatCoV HKU9. We were unable to obtain barcoding sequences for the three CoV-positive feces samples (GenBank: PQ305355, PQ305356, PQ305357); however, based on our observations, the cave where these samples were collected predominantly hosts Taphozous sp. bats.

Maximum likelihood phylogenetic trees of partial RdRp gene with different primers. (A) Phylogenetic tree of partial RdRp gene (aligned length 295 bp) sequenced using primers published by Quan et al.47. (B) Phylogenetic tree of partial RdRp gene (aligned length 389 bp) sequenced using primers published by Watanabe et al.48. Multiple sequence alignments were performed using the online Multiple alignment program for amino acid or nucleotide sequences (MAFFT Version 7, https://mafft.cbrc.jp/alignment/server/index.html). The sequences detected in Cambodia were marked by black triangles for strains in this study. The tree was built using the maximum likelihood method with the best-fitting nucleotide substitution model selected by the ModelFinder application in IQ-TREE. The robustness of nodes was assessed with 1000 bootstrap replicates. Bootstrap values < 70 are not shown. Scale bar indicates nucleotide substitutions per site. The GenBank accession numbers of CoV detected in this study and included in the phylogenetic analysis are available in supplemental file 6.
Discussion
We evaluated the prevalence and diversity of CoVs among a wide variety of wild animal species in two rural areas of Cambodia where rich biodiversity, significant environmental changes, wildlife trade and consumption have been documented. A total of 2,608 specimens were collected from at least 15 different genera and 25 species of wild animals between August 2020 and February 2022.
Molecular assays revealed an overall prevalence of 2.0% for CoVs, which were only found in rodents and bats. Rodentia and Chiroptera orders make up the majority of living mammals on earth55, and account for many of the reservoirs of zoonotic pathogens56. In our investigation, CoV RNA was identified in 3.3% of bats sampled and 3.4% among horseshoe bats. In a study conducted between 2010 and 2013 in Cambodia, Lacroix et al. found an overall CoV prevalence of 5.7% (61/1,059) among bats sampled and 1.9% (1/52) among horseshoe bats57. This higher CoV prevalence among Rhinolophus bats can be explained by our sampling method, targeting habitats of these bat species (i.e.: karstic caves and surrounding forests) but also by our detection methods. We combined detection of CoV RNA using two conventional PCRs and a real-time PCR, this latest being able to detect specifically SARS-CoV-2-like viruses, while the previous study used only one conventional PCR system. We observed differences in the capacity of our conventional systems to detect CoV (“Quan” versus “Watanabe” protocol), in line with a recent comparative performance study showing a higher sensitivity of “Quan” protocol58. However, all SARS-CoV-2 related RNA were detected by both real-time and “Watanabe” protocols, while only four by the “Quan” system. Our study confirms the necessity to combine several molecular detection systems to capture a large picture of CoV diversity among bats. In our study, partial RdRp sequences only were used in phylogenetic analysis, which might hamper our results interpretations. However, this marker remains valuable in detecting and classifying CoVs as it exhibits lower susceptibility to recombination compared to other regions of CoV genomes59. A full genome sequencing approach is underway to characterize these viruses and assess the actual relationship of these strains to SARS-CoV-2.
SARS-CoV-2-related viruses similar to those previously registered at the same sampling sites in Stung Treng in 201022 were detected among R. shameli bats in 2020 (n = 1) and 2021 (n = 8). Further characterization through full genome sequencing approaches of viruses detected in 2020 and 2021 is ongoing and may shed light on the evolution of viruses in bat populations in this area. Seasonal patterns of CoVs circulation in bats have been suggested, linked to parturition, lactation and occurrence of juvenile bats41,60,61,62. Reproductive patterns of horseshoe bats remain not fully characterized, but is occurring between March (gestation) and July (emergence of juveniles). As we detected SARS-CoV-2-related viruses during almost every capture session (October 2020 (n = 1), March 2021 (n = 1), December 2021 (n = 7)) some of which were outside of the aforementioned periods, this suggests a low but continuous viral circulation among these bat populations. We cannot assess potential seasonality of CoVs and/or SARS-CoV-2-related viruses in bats from our study as suggested by other authors as our sampling timeline only allow to capture a cross-sectional picture of CoVs circulation. However, the ongoing detection of such viruses in this area indicates the necessity of regular and systematic sampling, not just to assess the viral diversity but to also monitor any changes that could presage risk for human populations. During our study, we could not ensure that bats were not recaptured either within or between sampling sessions. This could introduce a potential bias in our interpretation of bat population dynamics at sampling sites. Regarding the detection of SARS-CoV-2-related viruses however, it is highly unlikely that the same viruses were identified from repeated sampling of the same individuals. While the relatively short sequences displayed high nucleotide similarity, this observation aligns with findings from previous studies, such as those reported by Delaune et al.22, where identical sequences on RdRp gene were identified from distinct bats sampled within the same area. Furthermore, the sex, reproductive and standard morphological characteristics of all bats sampled were carefully recorded and collectively indicate that the SARS-CoV-2-related virus-positive bats were distinct individuals.
Interestingly, two SARS-CoV-2-like viruses were detected from a parous female of T. melanopogon (n = 1) and a juvenile female of M. spasma (n = 1). To our knowledge, this is the first report of these species harboring a SARS-like virus. Wacharapluesadee et al. in Thailand identified T. melanopogon in Thailand as a reservoir for CoV with a prevalence of 1.6% (2/123) for HKU7- and HKU10-related alpha-CoV24. Very few studies have detected CoV among megadermatid bats. None of the M. spasma previously sampled in Cambodia were positive for CoV RNA41,57 and in Thailand, one M. lyra (now reclassified as Lyroderma lyra) was identified as a reservoir for HKU-10-related virus24. A review of CoV surveillance among African bats highlighted detections of AlphaCoV in the Megadermatidae63. T. melanopogon and M. spasma species are known to dwell in caves and artificial structures in Southeast Asia34 and the possibility that these co-roost with other species such as Rhinolophus in our study area cannot be ruled out64. The likelihood of virus transmission is not only higher among bats that co-roost in cave compared to species that roost elsewhere65 but can also be increased by higher population density of bats in caves and tendency to roost in clusters66. Cross-species transmission of CoVs among bats has already been documented67 however, because such viruses have only been detected in horseshoe bats so far, we cannot confirm at this stage the detection of SARS-CoV-2-like in these two bat species. Laboratory cross-contamination cannot be ruled out though all procedures in place at the Virology Unit of IPC were scrupulously followed. Further experiments are undergoing to investigate the presence of ACE2 receptor and their usage by these SARS-CoV-2-like viruses. Additional detection of such viruses in these bat species could help confirming these preliminary results. In our study, the phylogenetic analysis revealed closely related Embecovirus sequences from different bat species, including Rousettus, Taphozous and insectivorous bats. Although barcoding could not be retrieved for the feces samples labeled as ‘insectivorous,’ field observations during sample collection indicated that the cave predominantly hosted Taphozous sp. bats. Additionally, this sampling site is located near another cave housing a large colony of Rousettus bats, where feces samples were also collected and found positive for HKU9-related CoVs. While our study cannot confirm direct co-roosting or interactions between these species, the phylogenetic similarity of the detected sequences provides indirect evidence supporting the hypothesis of inter-species transmission. HKU10-related CoVs were detected in two different bat species, R. shameli and H. Pomona, captured either from the same location or from different sites. These findings are consistent with previous observations in Cambodia and Thailand, where such viruses were identified in bats from divergent families24,57. Interestingly, our HKU10-related CoVs share similarities with those found in Rousettus in China, where interspecies transmission between bats of different suborders was demonstrated68.
Bats have been identified as natural reservoir hosts for several emerging viruses of public health and veterinary importance6. In 2016, a significant outbreak of pig mortality in China was caused by a novel HKU2-related bat CoV, the swine acute diarrhea syndrome CoV (SADS-CoV) or swine enteric alphacoronavirus (SeACoV)7. Rhinolophus bats were identified as the main reservoir for this new CoV. In our survey, viruses closely related to SADS-CoV were detected in seven samples from R. shameli, adding pieces of evidence that horseshoes bats may be a reservoir for HKU2-related viruses in Cambodia. While SADS-CoV infection in human have not been documented, the possibility of cross-species transmission cannot be overlooked as this virus, like other CoVs, has significant potential for transmission between different species69. Sampling from this study did not include domestic animals so future study should investigate the presence of such virus in swine in the area.
Rodents are hosts for many zoonotic pathogens including hantavirus, arenavirus and CoVs, among others70,71. Human CoV OC43 and HKU1 likely originated from rodents, which are also known to harbor additional alpha- and beta-CoVs72,73. In our study, CoV prevalence was 2.5% (CI95 0.1–4.9) among captured rodents and belonged only to betaCoV from lineage A. Similar findings have been made in Malaysian Borneo74 and southern Vietnam75, where predominantly Rattus animals had detectable CoV RNA, and only betaCoV were described. In our study, all CoV detected among R. exulans (n = 3) were related to Parker’s rat CoV (PRC), known to be endemic in rats. Additionally, an unidentified Beta-CoV closely related to RatCoV Longquan was detected in a Rattus sp. trapped inside a bat cave in Stung Treng, where CoVs were also identified in bats. While the rodent and bat CoVs belonged to distinct Beta-CoVs sub-lineages (A and D, respectively), the ecological setting of the caves, where diverse groups such as bats and rodents coexist, warrants further investigation into the potential for pathogen spill over between species. The detection of rodent CoV RNA in M. musculus feces also inside a bat cave reinforces the importance of studying interactions between sympatric wildlife species and the role of shared habitats in facilitating viral circulation or exchange.
Through a collaborative approach with conservation NGO, private institutions and governmental agencies, we were able to include in our sampling effort specimen from, among others, Viverridae, Felidae and Mustelidae families, known to be susceptible to SARS-CoV-2 infection22 and potential intermediary hosts for transmission of CoVs to humans. No CoVs were detected among wildlife feces collected in Mondulkiri province, even in bat feces collected under tree roosts. Low sampling sizes for bat feces in Mondulkiri (n = 171, vs. n = 1,070 in Stung Treng) and other animal taxa may explain such results. In addition, one can assume the environmental conditions for feces preservation inside a tropical forest may not be optimal for CoV RNA detection. However, we demonstrate here the feasibility of a passive sample collection system as an alternative to live animal capture for pathogen surveillance among wildlife based on collaborative efforts between virologists and conservationists. This passive sampling can be equally useful to get samples from wildlife taxa that are difficult to find and to capture in the forested areas. Thanks to their expertise and appropriate training, rangers from government agencies and conservation NGOs were able to safely collect fresh feces from targeted taxa during their patrol activities. This type of collaboration could greatly help in future efforts to collect sample from various wildlife species to better understand not only the extend of CoV reservoirs but potential bridge species.
Preparedness and response to pandemics have taken a center stage over the past two decades, and beyond improving our understanding of zoonotic risks posed by wildlife, actions to reduce the risk of pandemics are still required. Major threats to bat populations across the world are human related, and mainly include logging, agriculture intensification, bat hunting and consumption, and human intrusions to bat habitats76. Conservation of bat habitats is not merely an ecological concern but also a matter of public health, and reducing human-bat interactions can mitigate the potential for spillover events. Karst caves in Stung Treng are regularly visited by human populations34 and the environment of the area is changing significantly at a landscape level35. Through understanding where and in which species coronaviruses are prevalent, targeted interventions can be designed to protect both the bats and the human communities that live nearby.
Conclusion
The identification of both alpha- and betacoronaviruses in karst caves in Stung Treng accentuates the necessity for ongoing surveillance and monitoring. Additionally, the discovery of new bat species potentially carrying SARS-CoV-2-like viruses in Cambodia sheds light on the complex landscape of bat-borne coronaviruses in a region where ecological and human factors converge. Studying bat ecology to understand the co-roosting behaviour and the risk of virus sharing among bat species is warranted. The findings of our research could be integrated into broader wildlife management strategies in Cambodia. Though capture and sampling of bats remain necessary to understand virus-host interactions, non-invasive sampling strategies could be employed in CoVs surveillance as a proxy for what is circulating among bats and other wildlife, helping reducing both the risk of disturbance of animal populations and risks of pathogen transmission to humans.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information. Sequence data have been deposited in GenBank and are provided in Supplemental file 6, along with reference sequences used in this study.
References
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable Bat origin. Nature 579 (7798), 270–273 (2020).
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C. & Garry, R. F. The proximal origin of SARS-CoV-2. Nat. Med. 26 (4), 450–452 (2020).
Islam, A. et al. Evolutionary dynamics and epidemiology of endemic and emerging coronaviruses in humans, domestic animals, and wildlife. Viruses 13 (10), 1908 (2021).
Ge, X. Y. et al. Isolation and characterization of a Bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503 (7477), 535–538 (2013).
Lau, S. K. P. et al. Genetic characterization of betacoronavirus lineage C viruses in Bats reveals marked sequence divergence in the Spike protein of pipistrellus Bat coronavirus HKU5 in Japanese pipistrelle: implications for the origin of the novel middle East respiratory syndrome coronavirus. J. Virol. 87 (15), 8638–8650 (2013).
Letko, M., Seifert, S. N., Olival, K. J., Plowright, R. K. & Munster, V. J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 18 (8), 461–471 (2020).
Zhou, P. et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of Bat origin. Nature 556 (7700), 255–258 (2018).
Schindell, B. G., Allardice, M., McBride, J. A. M., Dennehy, B. & Kindrachuk, J. SARS-CoV-2 and the missing link of intermediate hosts in viral emergence—What we can learn from other betacoronaviruses. Front. Virol. https://doi.org/10.3389/fviro.2022.875213 (2022).
Cui, J., Li, F. & Shi, Z. L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17 (3), 181–192 (2019).
Ye, Z. W. et al. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 16 (10), 1686–1697 (2020).
Forni, D., Cagliani, R., Clerici, M. & Sironi, M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 25 (1), 35–48 (2017).
MacLean, O. A. et al. Natural selection in the evolution of SARS-CoV-2 in bats created a generalist virus and highly capable human pathogen. PLoS Biol. 19 (3), e3001115 (2021).
Allen, T. et al. Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 8 (1), 1124 (2017).
Gibb, R. et al. Zoonotic host diversity increases in human-dominated ecosystems. Nature 584 (7821), 398–402 (2020).
Jones, B. A. et al. Zoonosis emergence linked to agricultural intensification and environmental change. Proc. Natl. Acad. Sci. USA 110 (21), 8399–8404 (2013).
Estoque, R. C. et al. The future of Southeast Asia’s forests. Nat. Commun. 10 (1), 1829 (2019).
Latinne, A. et al. One health surveillance highlights circulation of viruses with zoonotic potential in bats, pigs, and humans in Viet Nam. Viruses 15 (3), 790 (2023).
Cantlay, J. C., Ingram, D. J. & Meredith, A. L. A review of zoonotic infection risks associated with the wild meat trade in Malaysia. Ecohealth 14 (2), 361–388 (2017).
Chomel, B. B., Belotto, A. & Meslin, F. X. Wildlife, exotic pets, and emerging zoonoses. Emerg. Infect. Dis. J. CDC 13 (1), https://wwwnc.cdc.gov/eid/article/13/1/06-0480_article (2007).
Greatorex, Z. F. et al. Wildlife trade and human health in Lao PDR: an assessment of the zoonotic disease risk in markets. PLoS One 11 (3), e0150666 (2016).
Huong, N. Q. et al. Coronavirus testing indicates transmission risk increases along wildlife supply chains for human consumption in Viet Nam, 2013–2014. PLoS One 15 (8), e0237129 (2020).
Delaune, D. et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat. Commun. 12, 6563 (2021).
Temmam, S. et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 604 (7905), 330 (2022).
Wacharapluesadee, S. et al. Diversity of coronavirus in bats from Eastern Thailand. Virol. J. 12. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4416284/ (2015).
Zhou, H. et al. A novel Bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the Spike protein. Curr. Biol. 30 (11), 2196–2203e3 (2020).
Zhou, H. et al. Identification of novel Bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 184 (17), 4380–4391e14 (2021).
Gray, T. et al. Understanding and solving the South-East Asian snaring crisis. (2021).
Nielsen, M. R., Meilby, H., Smith-Hall, C., Pouliot, M. & Treue, T. The importance of wild meat in the global South. Ecol. Econ. 146, 696–705 (2018).
TRAFFIC. What’s driving the wildlife trade? A review of expert opinion on economic and social drivers of the wildlife trade and trade control efforts in Cambodia, Indonesia, Lao PDR and Vietnam. East Asia and Pacific region sustainable development discussion papers. East Asia and Pacific region sustainable development department. https://www.traffic.org/site/assets/files/5435/whats-driving-wildlife-trade.pdf (2008).
CIRAD. CIRAD. ZooCov. https://www.cirad.fr/en/worldwide/cirad-worldwide/projects/zoocov-project (2023).
Pacheco, P. et al. Deforestation fronts: drivers and responses in a changing world. WWF Gland Switz. (2021).
Singh, R., Boonratana, R., Bezuijen, M. & Phonvisay, M. Trade in natural resources in Stung Treng Province, Cambodia: An assessment of the wildlife trade - Wildlife Trade Report from TRAFFIC. 2006. https://www.traffic.org/publications/reports/trading-goods-in-cambodia/
Walston, J., Davidson, P. & Soriyun, M. A wildlife survey of Southern Mondulkiri Province, Cambodia. (2000).
Furey, N. M., Cappelle, J. & Racey, P. A. The conservation status of Cambodian cave bats. Laumanns M ed int speleol Proj Cambodia 2016 prov Stoeng Treng Kampong Speu Banteay meanchey BAttambang. 82–95. (2016).
Kong, R. et al. Understanding the drivers of deforestation and agricultural transformations in the Northwestern uplands of Cambodia. Appl. Geogr. 102, 84–98 (2019).
Pauly, M. & Tosteson, J. Safeguarding natural forests through the voluntary REDD + scheme. Nat. Plants. 8 (8), 861–866 (2022).
Buhler, D. et al. Rural Livelihood Strategies in Cambodia: Evidence from a Household Survey in Stung Treng (Zentrum für Entwicklungsforschung Center for Development Research, Univeristy of Bonn, 2015).
Sikes, R. S. Guidelines of the American society of mammalogists for the use of wild mammals in research and education. J. Mammal. 97 (3), 663–88. (2016).
American Veterinary Medical Association. AVMA guidelines for the euthanasia of animals. https://www.avma.org/resources-tools/avma-policies/avma-guidelines-euthanasia-animals
Percie du Sert, N. et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. Br. J. Pharmacol. 177 (16), 3617–3624 (2020).
Cappelle, J. et al. Longitudinal monitoring in Cambodia suggests higher circulation of alpha and betacoronaviruses in juvenile and immature bats of three species. Sci. Rep. 11 (1), 24145 (2021).
Francis, C. M. A Guide to the Mammals of Southeast Asia. https://books.google.com.kh/books?id=Jo3fGQAACAAJ (Princeton University Press, 2008).
Herbreteau, V. et al. Protocols for field and laboratory rodent studies. https://hal.ird.fr/ird-00714514 (2011).
Elephant Valley Project. Elephant Valley Project. https://elephantvalleyproject.org/ (2006).
BirdLife International. Cambodia - BirdLife International Cambodia Programme. https://www.birdlife.org/partners/cambodia-birdlife-international-cambodia-programme/ (2021).
Jahoo | Ecotourism. Gibbon Conservation and Research. https://gibbon.life/ (2020).
Quan, P. L. et al. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. mBio 1 (4). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2975989/ (2010).
Watanabe, S. et al. Bat coronaviruses and experimental infection of bats, the Philippines. Emerg. Infect. Dis. 16 (8), 1217–1223 (2010).
Corman, V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 25 (3), 2000045 (2020).
Townzen, J. S., Brower, A. V. Z. & Judd, D. D. Identification of mosquito bloodmeals using mitochondrial cytochrome oxidase subunit I and cytochrome B gene sequences. Med. Vet. Entomol. 22 (4), 386–393 (2008).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780 (2013).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274 (2015).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35 (2), 518–522 (2018).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14 (6), 587–589 (2017).
Wilson, D. E. & Reeder, D. M. Mammal Species of the World: A Taxonomic and Geographic Reference 2201 (JHU, 2005).
Olival, K. J. et al. Host and viral traits predict zoonotic spillover from mammals. Nature 546 (7660), 646–650 (2017).
Lacroix, A. et al. Genetic diversity of coronaviruses in bats in Lao PDR and Cambodia. Infect. Genet. Evol. 48, 10–18 (2017).
Wacharapluesadee, S. et al. Comparative performance in the detection of four coronavirus genera from human, animal, and environmental specimens. Viruses 16 (4), 534 (2024).
Gouilh, M. A. et al. SARS-Coronavirus ancestor’s foot-prints in South-East Asian Bat colonies and the refuge theory. Infect. Genet. Evol. 11 (7), 1690–1702 (2011).
Chidoti, V. et al. Longitudinal survey of coronavirus circulation and diversity in insectivorous Bat colonies in Zimbabwe. Viruses 14 (4), 781 (2022).
Geldenhuys, M. et al. Viral maintenance and excretion dynamics of coronaviruses within an Egyptian rousette fruit Bat maternal colony: considerations for spillover. Sci. Rep. 13 (1), 15829 (2023).
Joffrin, L. et al. Seasonality of coronavirus shedding in tropical bats. R Soc. Open. Sci. 9 (2), 211600 (2022).
Geldenhuys, M. et al. Overview of Bat and wildlife coronavirus surveillance in Africa: A framework for global investigations. Viruses 13 (5), 936 (2021).
Mainstreaming Karst Biodiversity Conservation into Policies, Plans, and Business Practices in Myanmar (Harrison Institute, 2015).
Willoughby, A. R., Phelps, K. L., Consortium, P. R. E. D. I. C. T. & Olival, K. J. A comparative analysis of viral richness and viral sharing in cave-roosting bats. Diversity 9 (3), 35 (2017).
Calisher, C. H., Childs, J. E., Field, H. E., Holmes, K. V. & Schountz, T. Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 19 (3), 531–545 (2006).
Latinne, A. et al. Origin and cross-species transmission of Bat coronaviruses in China. Nat. Commun. 11 (1), 4235 (2020).
Lau, S. K. P. et al. Recent transmission of a novel alphacoronavirus, Bat coronavirus HKU10, from Leschenault’s rousettes to Pomona leaf-nosed Bats: first evidence of interspecies transmission of coronavirus between Bats of different suborders. J. Virol. 86 (21), 11906–11918 (2012).
Mei, X. Q. et al. First evidence that an emerging mammalian alphacoronavirus is able to infect an avian species. Transbound. Emerg. Dis. 69 (5), e2006–e2019 (2022).
Han, B. A., Kramer, A. M. & Drake, J. M. Global patterns of zoonotic disease in mammals. Trends Parasitol. 32 (7), 565–577 (2016).
Meerburg, B. G., Singleton, G. R. & Kijlstra, A. Rodent-borne diseases and their risks for public health. Crit. Rev. Microbiol. 35 (3), 221–270 (2009).
Ge, X. Y. et al. Detection of alpha- and betacoronaviruses in rodents from Yunnan, China. Virol. J. 14, 98 (2017).
Wang, B. et al. Detection and characterization of three zoonotic viruses in wild rodents and shrews from Shenzhen City, China. Virol. Sin. 32 (4), 290–297 (2017).
Blasdell, K. R. et al. Rats and the City: implications of urbanization on zoonotic disease risk in Southeast Asia. Proc. Natl. Acad. Sci. 119 (39), e2112341119 (2022).
Berto, A. et al. Detection of potentially novel paramyxovirus and coronavirus viral RNA in bats and rats in the Mekong Delta region of Southern Viet Nam. Zoonoses Public. Health 65 (1), 30–42 (2018).
Frick, W. F., Kingston, T. & Flanders, J. A review of the major threats and challenges to global Bat conservation. Ann. N Y Acad. Sci. 1469 (1), 5–25 (2020).
Acknowledgements
We particularly acknowledge all relevant authorities from the Ministry of Agriculture, Forestry, and Fisheries and Ministry of Environment for their great support in facilitating this work. We are particularly thankful to the Department of Wildlife and Biodiversity under the Forestry Administration for their continuous support during field data collection. We are also grateful to Elephant Livelihood Initiative Environment Organization and Elephant Valley Project and in particular to Jemma Bullock for her help and support, to Jahoo community-based ecotourism project managed by World Hope International (WHI), the Mayura Wild Park, BirdLife International and Rising Phoenix CO. Ltd., the Wildlife Conservation Society (WCS) Cambodia and Regional Office, and the World Wildlife Fund (WWF) Cambodia for their great collaboration and support in the collection of environmental samples and wildlife individual specimens.
Funding
This work was carried out through the ZooCoV project funded by the French Research Agency (ANR-20-COVI-000), Région Occitanie, Pasteur Foundation Asia (https://umr-astre.cirad.fr/recherche/projets/zoocov) and RhinoKhoV project supported by MUSE (MUSE-16297). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
VC, VD, JC, NMF and JG contributed to the conception and design of the study. JG, VH, TH, NMF, CM, SN, SH and LK performed field data collection. JG, TPO, VH, TH, SN, SH, RL and LK performed laboratory analysis, JG and TPO performed data analysis, JG and VC performed the writing of the manuscript. All authors revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guillebaud, J., Ou, T.P., Hul, V. et al. Study of coronavirus diversity in wildlife in Northern Cambodia suggests continuous circulation of SARS-CoV-2-related viruses in bats. Sci Rep 15, 12628 (2025). https://doi.org/10.1038/s41598-025-92475-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-92475-x
