
Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19

 , ,
, ,
Abstract
:1. Introduction
2. Inflammation and Platelet Activation
2.1. Endothelial Dysfunction and Platelet Activation
2.2. Platelet–Neutrophil Interactions in Atherosclerosis
2.3. The Role of Myeloperoxidase and Platelet Toll-Like Receptors
2.4. Cytokines in Platelet Activation
2.5. NLRP3 Inflammasome
2.6. Platelet Factor-4
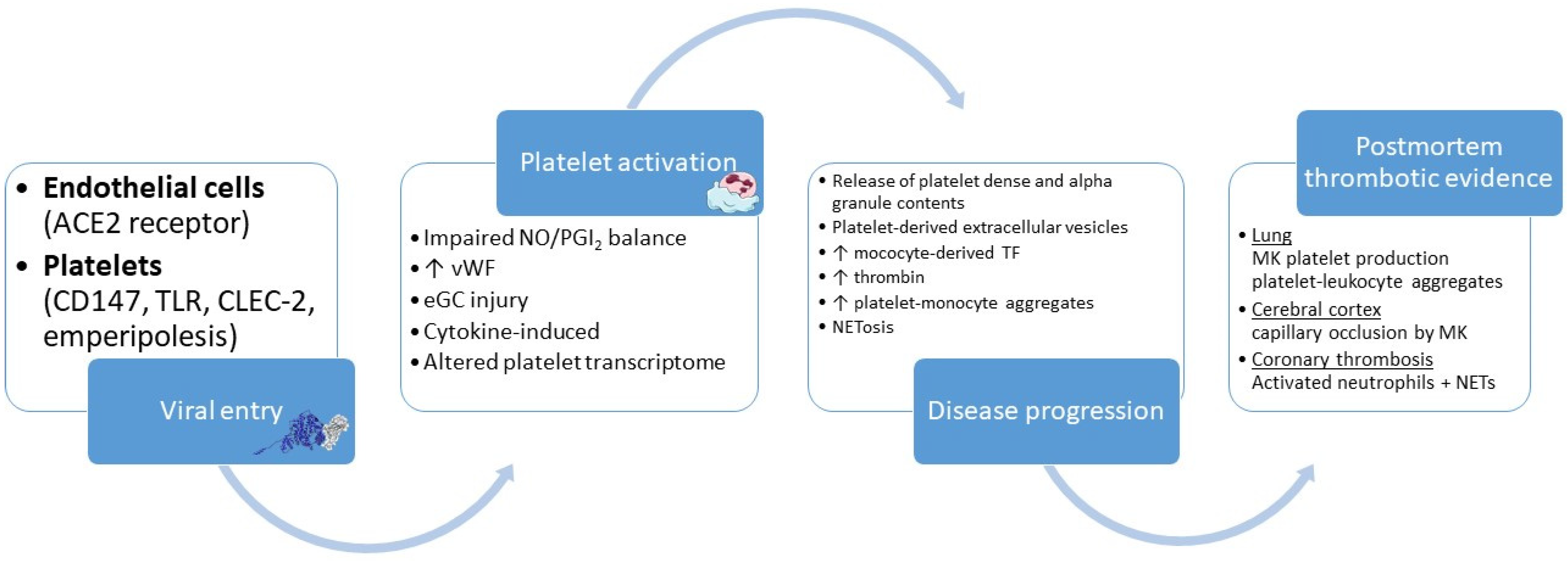
3. Inflammation and Platelet Activation in Coronavirus Disease-19
4. Therapeutic Implications
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oikonomou, E.; Vogiatzi, G.; Lazaros, G.; Tsalamandris, S.; Goliopoulou, A.; Mystakidou, V.; Theofilis, P.; Christoforatou, E.; Chasikidis, C.; Tousoulis, D. Relationship of depressive symptoms with arterial stiffness and carotid atherosclerotic burden in the Corinthia study. QJM Mon. J. Assoc. Physicians 2020, 113, 633–642. [Google Scholar] [CrossRef]
- Oikonomou, E.; Lazaros, G.; Mystakidi, V.C.; Papaioannou, N.; Theofilis, P.; Vogiatzi, G.; Chasikidis, C.; Fountoulakis, P.; Papakostantinou, M.A.; Assimakopoulos, M.N.; et al. The association of air pollutants exposure with subclinical inflammation and carotid atherosclerosis. Int. J. Cardiol. 2021, 342, 108–114. [Google Scholar] [CrossRef]
- Oikonomou, E.; Lazaros, G.; Christoforatou, E.; Chasikidis, C.; Vavouranaki, G.; Vogiatzi, G.; Papamikroulis, G.A.; Tsalamandris, S.; Gergiopoulos, G.; Mazaris, S.; et al. Breakfast association with arterial stiffness and carotid atherosclerotic burden. Insights from the ‘Corinthia’ study. Nutr. Metab. Cardiovasc. Dis. NMCD 2019, 29, 744–750. [Google Scholar] [CrossRef]
- Oikonomou, E.; Theofilis, P.; Vogiatzi, G.; Lazaros, G.; Tsalamandris, S.; Mystakidi, V.C.; Goliopoulou, A.; Anastasiou, M.; Fountoulakis, P.; Chasikidis, C.; et al. The impact of sleeping duration on atherosclerosis in the community: Insights from the Corinthia study. Sleep Breath. 2021. [Google Scholar] [CrossRef]
- Majithia, A.; Bhatt, D.L. Novel Antiplatelet Therapies for Atherothrombotic Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Siasos, G.; Skotsimara, G.; Oikonomou, E.; Sagris, M.; Vasiliki-Chara, M.; Bletsa, E.; Stampouloglou, P.; Theofilis, P.; Charalampous, G.; Tousoulis, D. Antithrombotic Treatment in Diabetes Mellitus: A Review of the Literature about Antiplatelet and Anticoagulation Strategies Used for Diabetic Patients in Primary and Secondary Prevention. Curr. Pharm. Des. 2020, 26, 2780–2788. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef] [PubMed]
- Delgadillo, L.F.; Lomakina, E.B.; Kuebel, J.; Waugh, R.E. Changes in endothelial glycocalyx layer protective ability after inflammatory stimulus. Am. J. Physiol. Cell Physiol. 2021, 320, C216–C224. [Google Scholar] [CrossRef] [PubMed]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The glycocalyx: A novel diagnostic and therapeutic target in sepsis. Crit. Care 2019, 23, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, B.F.; Jacob, M.; Leipert, S.; Salmon, A.H.; Chappell, D. Degradation of the endothelial glycocalyx in clinical settings: Searching for the sheddases. Br. J. Clin. Pharmacol. 2015, 80, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Lukasz, A.; Hillgruber, C.; Oberleithner, H.; Kusche-Vihrog, K.; Pavenstadt, H.; Rovas, A.; Hesse, B.; Goerge, T.; Kumpers, P. Endothelial glycocalyx breakdown is mediated by angiopoietin-2. Cardiovasc. Res. 2017, 113, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Echtermeyer, F.; Alozie, A.; Brands, K.; Buddecke, E. Plasmin- and thrombin-accelerated shedding of syndecan-4 ectodomain generates cleavage sites at Lys(114)-Arg(115) and Lys(129)-Val(130) bonds. J. Biol. Chem. 2005, 280, 34441–34446. [Google Scholar] [CrossRef] [Green Version]
- Britten, M.W.; Lumers, L.; Tominaga, K.; Peters, J.; Dirkmann, D. Glycocalyx components affect platelet function, whole blood coagulation, and fibrinolysis: An in vitro study suggesting a link to trauma-induced coagulopathy. BMC Anesthesiol. 2021, 21, 83. [Google Scholar] [CrossRef]
- Reitsma, S.; Oude Egbrink, M.G.; Heijnen, V.V.; Megens, R.T.; Engels, W.; Vink, H.; Slaaf, D.W.; van Zandvoort, M.A. Endothelial glycocalyx thickness and platelet-vessel wall interactions during atherogenesis. Thromb. Haemost. 2011, 106, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Constantinescu, A.A.; Vink, H.; Spaan, J.A. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1541–1547. [Google Scholar] [CrossRef] [Green Version]
- Becker, B.F.; Chappell, D.; Bruegger, D.; Annecke, T.; Jacob, M. Therapeutic strategies targeting the endothelial glycocalyx: Acute deficits, but great potential. Cardiovasc. Res. 2010, 87, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Dogne, S.; Flamion, B.; Caron, N. Endothelial Glycocalyx as a Shield Against Diabetic Vascular Complications: Involvement of Hyaluronan and Hyaluronidases. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1427–1439. [Google Scholar] [CrossRef]
- Smolenski, A. Novel roles of cAMP/cGMP-dependent signaling in platelets. J. Thromb. Haemost. 2012, 10, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellion, B.T.; Ignarro, L.J.; Ohlstein, E.H.; Pontecorvo, E.G.; Hyman, A.L.; Kadowitz, P.J. Evidence for the inhibitory role of guanosine 3′, 5′-monophosphate in ADP-induced human platelet aggregation in the presence of nitric oxide and related vasodilators. Blood 1981, 57, 946–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigenthaler, M.; Ullrich, H.; Geiger, J.; Horstrup, K.; Honig-Liedl, P.; Wiebecke, D.; Walter, U. Defective nitrovasodilator-stimulated protein phosphorylation and calcium regulation in cGMP-dependent protein kinase-deficient human platelets of chronic myelocytic leukemia. J. Biol. Chem. 1993, 268, 13526–13531. [Google Scholar] [CrossRef]
- Massberg, S.; Sausbier, M.; Klatt, P.; Bauer, M.; Pfeifer, A.; Siess, W.; Fassler, R.; Ruth, P.; Krombach, F.; Hofmann, F. Increased adhesion and aggregation of platelets lacking cyclic guanosine 3′,5′-monophosphate kinase I. J. Exp. Med. 1999, 189, 1255–1264. [Google Scholar] [CrossRef]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar]
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef]
- Freedman, J.E.; Loscalzo, J.; Barnard, M.R.; Alpert, C.; Keaney, J.F.; Michelson, A.D. Nitric oxide released from activated platelets inhibits platelet recruitment. J. Clin. Investig. 1997, 100, 350–356. [Google Scholar] [CrossRef]
- Berkels, R.; Bertsch, A.; Zuther, T.; Dhein, S.; Stockklauser, K.; Rosen, P.; Rosen, R. Evidence for a NO synthase in porcine platelets which is stimulated during activation/aggregation. Eur. J. Haematol. 1997, 58, 307–313. [Google Scholar] [CrossRef]
- Malinski, T.; Radomski, M.W.; Taha, Z.; Moncada, S. Direct electrochemical measurement of nitric oxide released from human platelets. Biochem. Biophys. Res. Commun. 1993, 194, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E.; Sauter, R.; Battinelli, E.M.; Ault, K.; Knowles, C.; Huang, P.L.; Loscalzo, J. Deficient platelet-derived nitric oxide and enhanced hemostasis in mice lacking the NOSIII gene. Circ. Res. 1999, 84, 1416–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momi, S.; Caracchini, R.; Falcinelli, E.; Evangelista, S.; Gresele, P. Stimulation of platelet nitric oxide production by nebivolol prevents thrombosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, U.R.; Walter, U.; Eigenthaler, M. Taming platelets with cyclic nucleotides. Biochem. Pharmacol. 2001, 62, 1153–1161. [Google Scholar] [CrossRef]
- Ramadan, F.M.; Upchurch, G.R., Jr.; Keagy, B.A.; Johnson, G., Jr. Endothelial cell thromboxane production and its inhibition by a calcium-channel blocker. Ann. Thorac. Surg. 1990, 49, 916–919. [Google Scholar] [CrossRef]
- Ooi, B.S.; Cohen, D.J.; Chang, T.H.; Tian, Y.; Papademetrious, V. Stimulation of endothelial cell production of vasoconstrictive substances by hypertensive sera. Am. J. Hypertens. 1998, 11, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Wagner, D.D.; Marder, V.J. Biosynthesis of von Willebrand protein by human endothelial cells: Processing steps and their intracellular localization. J. Cell Biol. 1984, 99, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.D.; Atkinson, T.M.; Lindner, J.R. Platelets and von Willebrand factor in atherogenesis. Blood 2017, 129, 1415–1419. [Google Scholar] [CrossRef]
- Dong, J.F.; Moake, J.L.; Nolasco, L.; Bernardo, A.; Arceneaux, W.; Shrimpton, C.N.; Schade, A.J.; McIntire, L.V.; Fujikawa, K.; Lopez, J.A. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002, 100, 4033–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.J.; Niiya, M.; Zheng, X.W.; Shang, D.Z.; Zheng, X.L. Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J. Thromb. Haemost. 2008, 6, 1233–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Fu, X.; Wang, Y.; Ling, M.; McMullen, B.; Kulman, J.; Chung, D.W.; Lopez, J.A. Oxidative modification of von Willebrand factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood 2010, 115, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Okhota, S.; Melnikov, I.; Avtaeva, Y.; Kozlov, S.; Gabbasov, Z. Shear Stress-Induced Activation of von Willebrand Factor and Cardiovascular Pathology. Int. J. Mol. Sci. 2020, 21, 7804. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Muller, M.A.; Varlamov, O.; Tavori, H.; Packwood, W.; Mueller, P.A.; Xie, A.; Ruggeri, Z.; Chung, D.; Lopez, J.A.; et al. Proteolysis of Von Willebrand Factor Influences Inflammatory Endothelial Activation and Vascular Compliance in Atherosclerosis. JACC Basic Transl. Sci. 2020, 5, 1017–1028. [Google Scholar] [CrossRef]
- Zucoloto, A.Z.; Jenne, C.N. Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front. Cardiovasc. Med. 2019, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Rigg, R.A.; Healy, L.D.; Chu, T.T.; Ngo, A.T.P.; Mitrugno, A.; Zilberman-Rudenko, J.; Aslan, J.E.; Hinds, M.T.; Vecchiarelli, L.D.; Morgan, T.K.; et al. Protease-activated receptor 4 activity promotes platelet granule release and platelet-leukocyte interactions. Platelets 2019, 30, 126–135. [Google Scholar] [CrossRef]
- Seif, K.; Alidzanovic, L.; Tischler, B.; Ibrahim, N.; Zagrapan, B.; Rauscher, S.; Salzmann, M.; Hell, L.; Mauracher, L.M.; Budde, U.; et al. Neutrophil-Mediated Proteolysis of Thrombospondin-1 Promotes Platelet Adhesion and String Formation. Thromb. Haemost. 2018, 118, 2074–2085. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, E.; Leopoulou, M.; Theofilis, P.; Antonopoulos, A.S.; Siasos, G.; Latsios, G.; Mystakidi, V.C.; Antoniades, C.; Tousoulis, D. A link between inflammation and thrombosis in atherosclerotic cardiovascular diseases: Clinical and therapeutic implications. Atherosclerosis 2020, 309, 16–26. [Google Scholar] [CrossRef]
- Quinn, K.L.; Henriques, M.; Tabuchi, A.; Han, B.; Yang, H.; Cheng, W.E.; Tole, S.; Yu, H.; Luo, A.; Charbonney, E.; et al. Human neutrophil peptides mediate endothelial-monocyte interaction, foam cell formation, and platelet activation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2070–2079. [Google Scholar] [CrossRef] [Green Version]
- Horn, M.; Bertling, A.; Brodde, M.F.; Muller, A.; Roth, J.; Van Aken, H.; Jurk, K.; Heilmann, C.; Peters, G.; Kehrel, B.E. Human neutrophil alpha-defensins induce formation of fibrinogen and thrombospondin-1 amyloid-like structures and activate platelets via glycoprotein IIb/IIIa. J. Thromb. Haemost. 2012, 10, 647–661. [Google Scholar] [CrossRef]
- Kaiser, P.; Harenberg, J.; Walenga, J.M.; Huhle, G.; Giese, C.; Prechel, M.; Hoppensteadt, D.; Fareed, J. Effects of a heparin-binding protein on blood coagulation and platelet function. Semin. Thromb. Hemost. 2001, 27, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Faraday, N.; Schunke, K.; Saleem, S.; Fu, J.; Wang, B.; Zhang, J.; Morrell, C.; Dore, S. Cathepsin G-dependent modulation of platelet thrombus formation in vivo by blood neutrophils. PLoS ONE 2013, 8, e71447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruenster, M.; Kurz, A.R.; Chung, K.J.; Cao-Ehlker, X.; Bieber, S.; Nussbaum, C.F.; Bierschenk, S.; Eggersmann, T.K.; Rohwedder, I.; Heinig, K.; et al. Extracellular MRP8/14 is a regulator of beta2 integrin-dependent neutrophil slow rolling and adhesion. Nat. Commun. 2015, 6, 6915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santilli, F.; Paloscia, L.; Liani, R.; Di Nicola, M.; Di Marco, M.; Lattanzio, S.; La Barba, S.; Pascale, S.; Mascellanti, M.; Davi, G. Circulating myeloid-related protein-8/14 is related to thromboxane-dependent platelet activation in patients with acute coronary syndrome, with and without ongoing low-dose aspirin treatment. J. Am. Heart Assoc. 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Xiu, C.; Liu, M.; Lin, C.; Chen, H.; Bao, R.; Yang, S.; Yu, J. Platelet-neutrophil interaction aggravates vascular in fl ammation and promotes the progression of atherosclerosis by activating the TLR4/NF-kappaB pathway. J. Cell. Biochem. 2019, 120, 5612–5619. [Google Scholar] [CrossRef] [PubMed]
- Pircher, J.; Czermak, T.; Ehrlich, A.; Eberle, C.; Gaitzsch, E.; Margraf, A.; Grommes, J.; Saha, P.; Titova, A.; Ishikawa-Ankerhold, H.; et al. Cathelicidins prime platelets to mediate arterial thrombosis and tissue inflammation. Nat. Commun. 2018, 9, 1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef]
- Thakur, M.; Evans, B.; Schindewolf, M.; Baumgartner, I.; Doring, Y. Neutrophil Extracellular Traps Affecting Cardiovascular Health in Infectious and Inflammatory Diseases. Cells 2021, 10, 1689. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schaffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [Green Version]
- De Boer, O.J.; Li, X.; Teeling, P.; Mackaay, C.; Ploegmakers, H.J.; van der Loos, C.M.; Daemen, M.J.; de Winter, R.J.; van der Wal, A.C. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013, 109, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Riegger, J.; Byrne, R.A.; Joner, M.; Chandraratne, S.; Gershlick, A.H.; Ten Berg, J.M.; Adriaenssens, T.; Guagliumi, G.; Godschalk, T.C.; Neumann, F.J.; et al. Histopathological evaluation of thrombus in patients presenting with stent thrombosis. A multicenter European study: A report of the prevention of late stent thrombosis by an interdisciplinary global European effort consortium. Eur. Heart J. 2016, 37, 1538–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laridan, E.; Denorme, F.; Desender, L.; Francois, O.; Andersson, T.; Deckmyn, H.; Vanhoorelbeke, K.; De Meyer, S.F. Neutrophil extracellular traps in ischemic stroke thrombi. Ann. Neurol. 2017, 82, 223–232. [Google Scholar] [CrossRef]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, M.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Nussbaum, C.; Klinke, A.; Adam, M.; Baldus, S.; Sperandio, M. Myeloperoxidase: A leukocyte-derived protagonist of inflammation and cardiovascular disease. Antioxid. Redox Signal. 2013, 18, 692–713. [Google Scholar] [CrossRef]
- Baldus, S.; Heeschen, C.; Meinertz, T.; Zeiher, A.M.; Eiserich, J.P.; Munzel, T.; Simoons, M.L.; Hamm, C.W.; Investigators, C. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation 2003, 108, 1440–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kargapolova, Y.; Geissen, S.; Zheng, R.; Baldus, S.; Winkels, H.; Adam, M. The Enzymatic and Non-Enzymatic Function of Myeloperoxidase (MPO) in Inflammatory Communication. Antioxidants 2021, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Teng, N.; Maghzal, G.J.; Talib, J.; Rashid, I.; Lau, A.K.; Stocker, R. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep. Commun. Free Radic. Res. 2017, 22, 51–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolarova, H.; Klinke, A.; Kremserova, S.; Adam, M.; Pekarova, M.; Baldus, S.; Eiserich, J.P.; Kubala, L. Myeloperoxidase induces the priming of platelets. Free. Radic. Biol. Med. 2013, 61, 357–369. [Google Scholar] [CrossRef]
- Freedman, J.E.; Ting, B.; Hankin, B.; Loscalzo, J.; Keaney, J.F., Jr.; Vita, J.A. Impaired platelet production of nitric oxide predicts presence of acute coronary syndromes. Circulation 1998, 98, 1481–1486. [Google Scholar] [CrossRef]
- Gurses, K.M.; Kocyigit, D.; Yalcin, M.U.; Canpinar, H.; Oto, M.A.; Ozer, N.; Tokgozoglu, L.; Guc, D.; Aytemir, K. Enhanced Platelet Toll-like Receptor 2 and 4 Expression in Acute Coronary Syndrome and Stable Angina Pectoris. Am. J. Cardiol. 2015, 116, 1666–1671. [Google Scholar] [CrossRef]
- Hally, K.E.; La Flamme, A.C.; Larsen, P.D.; Harding, S.A. Platelet Toll-like receptor (TLR) expression and TLR-mediated platelet activation in acute myocardial infarction. Thromb. Res. 2017, 158, 8–15. [Google Scholar] [CrossRef]
- Rivadeneyra, L.; Carestia, A.; Etulain, J.; Pozner, R.G.; Fondevila, C.; Negrotto, S.; Schattner, M. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb. Res. 2014, 133, 235–243. [Google Scholar] [CrossRef]
- Kalvegren, H.; Skoglund, C.; Helldahl, C.; Lerm, M.; Grenegard, M.; Bengtsson, T. Toll-like receptor 2 stimulation of platelets is mediated by purinergic P2X1-dependent Ca2+ mobilisation, cyclooxygenase and purinergic P2Y1 and P2Y12 receptor activation. Thromb. Haemost. 2010, 103, 398–407. [Google Scholar] [CrossRef]
- Klarstrom Engstrom, K.; Brommesson, C.; Kalvegren, H.; Bengtsson, T. Toll like receptor 2/1 mediated platelet adhesion and activation on bacterial mimetic surfaces is dependent on src/Syk-signaling and purinergic receptor P2X1 and P2Y12 activation. Biointerphases 2014, 9, 041003. [Google Scholar] [CrossRef]
- Lopes Pires, M.E.; Clarke, S.R.; Marcondes, S.; Gibbins, J.M. Lipopolysaccharide potentiates platelet responses via toll-like receptor 4-stimulated Akt-Erk-PLA2 signalling. PLoS ONE 2017, 12, e0186981. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.L.; Svensson, M.; Morgelin, M.; Svanborg, C.; Tarr, P.I.; Mooney, J.C.; Watkins, S.L.; Johnson, R.; Karpman, D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 2006, 108, 167–176. [Google Scholar] [CrossRef] [Green Version]
- de Stoppelaar, S.F.; Claushuis, T.A.; Schaap, M.C.; Hou, B.; van der Poll, T.; Nieuwland, R.; van ‘t Veer, C. Toll-Like Receptor Signalling Is Not Involved in Platelet Response to Streptococcus pneumoniae In Vitro or In Vivo. PLoS ONE 2016, 11, e0156977. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, L.P.; Etulain, J.; Rivadeneyra, L.; Lapponi, M.J.; Centurion, M.; Cheng, K.; Yin, H.; Schattner, M. Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J. Thromb. Haemost. 2015, 13, 839–850. [Google Scholar] [CrossRef]
- Rex, S.; Beaulieu, L.M.; Perlman, D.H.; Vitseva, O.; Blair, P.S.; McComb, M.E.; Costello, C.E.; Freedman, J.E. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thromb. Haemost. 2009, 102, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 21, 6150. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Oikonomou, E.; Economou, E.K.; Crea, F.; Kaski, J.C. Inflammatory cytokines in atherosclerosis: Current therapeutic approaches. Eur. Heart J. 2016, 37, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Lin, A.; He, Q.Q.; Burgess, S.; Larsson, S.C. Circulating interleukins in relation to coronary artery disease, atrial fibrillation and ischemic stroke and its subtypes: A two-sample Mendelian randomization study. Int. J. Cardiol. 2020, 313, 99–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Tedgui, A.; Mallat, Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef] [Green Version]
- Regnault, V.; de Maistre, E.; Carteaux, J.P.; Gruel, Y.; Nguyen, P.; Tardy, B.; Lecompte, T. Platelet activation induced by human antibodies to interleukin-8. Blood 2003, 101, 1419–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, M.J.; Bester, J.; Pretorius, E. Interleukin-12 and its procoagulant effect on erythrocytes, platelets and fibrin(ogen): The lesser known side of inflammation. Br. J. Haematol. 2018, 180, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bester, J.; Pretorius, E. Effects of IL-1beta, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci. Rep. 2016, 6, 32188. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; Bester, J.; Pretorius, E. The inflammatory effects of TNF-alpha and complement component 3 on coagulation. Sci. Rep. 2018, 8, 1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfredi, A.A.; Baldini, M.; Camera, M.; Baldissera, E.; Brambilla, M.; Peretti, G.; Maseri, A.; Rovere-Querini, P.; Tremoli, E.; Sabbadini, M.G.; et al. Anti-TNFalpha agents curb platelet activation in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1511–1520. [Google Scholar] [CrossRef]
- Davizon-Castillo, P.; McMahon, B.; Aguila, S.; Bark, D.; Ashworth, K.; Allawzi, A.; Campbell, R.A.; Montenont, E.; Nemkov, T.; D’Alessandro, A.; et al. TNF-alpha-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood 2019, 134, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Folco, E.J.; Sukhova, G.K.; Quillard, T.; Libby, P. Moderate hypoxia potentiates interleukin-1beta production in activated human macrophages. Circ. Res. 2014, 115, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Lu, M.; Lin, T.Y.; Chen, Z.; Chen, G.; Wang, W.C.; Marin, T.; Shentu, T.P.; Wen, L.; Gongol, B.; et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation 2013, 128, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Monteiro, A.P.; Bozza, F.A.; Bozza, P.T. Inflammasome in platelets: Allying coagulation and inflammation in infectious and sterile diseases? Mediat. Inflamm. 2015, 2015, 435783. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; Narayanan, P.; Li, W.; Silverstein, R.L.; McIntyre, T.M. Lipopolysaccharide stimulates platelets through an IL-1beta autocrine loop. J. Immunol. 2013, 191, 5196–5203. [Google Scholar] [CrossRef] [Green Version]
- Murthy, P.; Durco, F.; Miller-Ocuin, J.L.; Takedai, T.; Shankar, S.; Liang, X.; Liu, X.; Cui, X.; Sachdev, U.; Rath, D.; et al. The NLRP3 inflammasome and bruton’s tyrosine kinase in platelets co-regulate platelet activation, aggregation, and in vitro thrombus formation. Biochem. Biophys. Res. Commun. 2017, 483, 230–236. [Google Scholar] [CrossRef]
- Vogel, S.; Kamimura, S.; Arora, T.; Smith, M.L.; Almeida, L.E.F.; Combs, C.A.; Thein, S.L.; Quezado, Z.M.N. NLRP3 inflammasome and bruton tyrosine kinase inhibition interferes with upregulated platelet aggregation and in vitro thrombus formation in sickle cell mice. Biochem. Biophys. Res. Commun. 2021, 555, 196–201. [Google Scholar] [CrossRef]
- Hottz, E.D.; Lopes, J.F.; Freitas, C.; Valls-de-Souza, R.; Oliveira, M.F.; Bozza, M.T.; Da Poian, A.T.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, F.A.; et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 2013, 122, 3405–3414. [Google Scholar] [CrossRef] [Green Version]
- Pitsilos, S.; Hunt, J.; Mohler, E.R.; Prabhakar, A.M.; Poncz, M.; Dawicki, J.; Khalapyan, T.Z.; Wolfe, M.L.; Fairman, R.; Mitchell, M.; et al. Platelet factor 4 localization in carotid atherosclerotic plaques: Correlation with clinical parameters. Thromb. Haemost. 2003, 90, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Aidoudi, S.; Bikfalvi, A. Interaction of PF4 (CXCL4) with the vasculature: A role in atherosclerosis and angiogenesis. Thromb. Haemost. 2010, 104, 941–948. [Google Scholar] [CrossRef]
- Gremmel, T.; Ay, C.; Riedl, J.; Kopp, C.W.; Eichelberger, B.; Koppensteiner, R.; Panzer, S. Platelet-specific markers are associated with monocyte-platelet aggregate formation and thrombin generation potential in advanced atherosclerosis. Thromb. Haemost. 2016, 115, 615–621. [Google Scholar] [CrossRef] [Green Version]
- Bhatnagar, P.; Lu, X.; Evans, M.K.; Laveist, T.A.; Zonderman, A.B.; Carter, D.L.; Arking, D.E.; Fletcher, C.A. Genetic variants in platelet factor 4 modulate inflammatory and platelet activation biomarkers. Circ. Cardiovasc. Genet. 2012, 5, 412–421. [Google Scholar] [CrossRef]
- Lord, M.S.; Cheng, B.; Farrugia, B.L.; McCarthy, S.; Whitelock, J.M. Platelet Factor 4 Binds to Vascular Proteoglycans and Controls Both Growth Factor Activities and Platelet Activation. J. Biol. Chem. 2017, 292, 4054–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Tsioufis, C.; Oikonomou, E.; Antoniades, C.; Crea, F.; Kaski, J.C.; Tousoulis, D. Inflammatory Mechanisms in COVID-19 and Atherosclerosis: Current Pharmaceutical Perspectives. Int. J. Mol. Sci. 2021, 22, 6607. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.; Aznaouridis, K.; Barbetseas, J.; Charalambous, G.; Gastouniotis, I.; Fotopoulos, V.; Gkini, K.P.; Katsivas, A.; Koudounis, G.; Koudounis, P.; et al. Hospital attendance and admission trends for cardiac diseases during the COVID-19 outbreak and lockdown in Greece. Public Health 2020, 187, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Lazaros, G.; Oikonomou, E.; Theofilis, P.; Theodoropoulou, A.; Triantafyllou, K.; Charitos, C.; Charalambous, G.; Papanikolaou, A.; Gastouniotis, I.; Siasos, G.; et al. The impact of COVID-19 pandemic on adult cardiac surgery procedures. Hell. J. Cardiol. 2021, 62, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.K.; Mainbourg, S.; Friggeri, A.; Bertoletti, L.; Douplat, M.; Dargaud, Y.; Grange, C.; Lobbes, H.; Provencher, S.; Lega, J.C. Arterial and venous thromboembolism in COVID-19: A study-level meta-analysis. Thorax 2021, 76, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Choudry, F.A.; Hamshere, S.M.; Rathod, K.S.; Akhtar, M.M.; Archbold, R.A.; Guttmann, O.P.; Woldman, S.; Jain, A.K.; Knight, C.J.; Baumbach, A.; et al. High Thrombus Burden in Patients With COVID-19 Presenting With ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2020, 76, 1168–1176. [Google Scholar] [CrossRef]
- Rodriguez-Leor, O.; Cid Alvarez, A.B.; Perez de Prado, A.; Rossello, X.; Ojeda, S.; Serrador, A.; Lopez-Palop, R.; Martin-Moreiras, J.; Rumoroso, J.R.; Cequier, A.; et al. In-hospital outcomes of COVID-19 ST-elevation myocardial infarction patients. EuroIntervention 2021, 16, 1426–1433. [Google Scholar] [CrossRef]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.M.; Charytan, D.M.; Gasmi, B.; et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine 2020, 24, 100434. [Google Scholar] [CrossRef]
- Portier, I.; Campbell, R.A. Role of Platelets in Detection and Regulation of Infection. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 70–78. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Khismatullin, R.R.; Ponomareva, A.A.; Nagaswami, C.; Ivaeva, R.A.; Montone, K.T.; Weisel, J.W.; Litvinov, R.I. Pathology of lung-specific thrombosis and inflammation in COVID-19. J. Thromb. Haemost. 2021. [Google Scholar] [CrossRef]
- Nauen, D.W.; Hooper, J.E.; Stewart, C.M.; Solomon, I.H. Assessing Brain Capillaries in Coronavirus Disease 2019. JAMA Neurol. 2021, 78, 760–762. [Google Scholar] [CrossRef]
- McMullen, P.; Smith, H.; Pytel, P. Entrapped Megakaryocytes in the Microvasculature of Brain Tissues are not Specific to COVID-19 but can be Seen Across a Spectrum of Acute Lung Injuries. J. Neuropathol. Exp. Neurol. 2021. [Google Scholar] [CrossRef]
- Johnson, J.E.; McGuone, D.; Xu, M.L.; Jane-Wit, D.; Mitchell, R.N.; Libby, P.; Pober, J.S. COVID Coronary Vascular Thrombosis: Correlation with Neutrophil but not Endothelial Activation. Am. J. Pathol. 2021. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Maquet, J.; Lafaurie, M.; Sommet, A.; Moulis, G. Thrombocytopenia is independently associated with poor outcome in patients hospitalized for COVID-19. Br. J. Haematol. 2020, 190, e276–e279. [Google Scholar] [CrossRef]
- Xu, P.; Zhou, Q.; Xu, J. Mechanism of thrombocytopenia in COVID-19 patients. Ann. Hematol. 2020, 99, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thachil, J.; Lisman, T. Pulmonary Megakaryocytes in Coronavirus Disease 2019 (COVID-19): Roles in Thrombi and Fibrosis. Semin. Thromb. Hemost. 2020, 46, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Q.; Wang, Y.; Wu, Y.; Xu, J.; Yu, Y.; Shang, Y. Thrombocytopenia and its association with mortality in patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Wool, G.D.; Miller, J.L. The Impact of COVID-19 Disease on Platelets and Coagulation. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2021, 88, 15–27. [Google Scholar] [CrossRef]
- Cohen, A.; Harari, E.; Cipok, M.; Laish-Farkash, A.; Bryk, G.; Yahud, E.; Sela, Y.; Lador, N.K.; Mann, T.; Mayo, A.; et al. Immature platelets in patients hospitalized with Covid-19. J. Thromb. Thrombolysis 2021, 51, 608–616. [Google Scholar] [CrossRef]
- Bongiovanni, D.; Santamaria, G.; Klug, M.; Santovito, D.; Felicetta, A.; Hristov, M.; von Scheidt, M.; Aslani, M.; Cibella, J.; Weber, C.; et al. Transcriptome Analysis of Reticulated Platelets Reveals a Prothrombotic Profile. Thromb. Haemost. 2019, 119, 1795–1806. [Google Scholar] [CrossRef]
- Tang, W.H.; Stitham, J.; Gleim, S.; Di Febbo, C.; Porreca, E.; Fava, C.; Tacconelli, S.; Capone, M.; Evangelista, V.; Levantesi, G.; et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J. Clin. Investig. 2011, 121, 4462–4476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barale, C.; Russo, I. Influence of Cardiometabolic Risk Factors on Platelet Function. Int. J. Mol. Sci. 2020, 21, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Qin, Z.; Liu, F.; Blair, R.; Wang, C.; Yang, H.; Mudd, J.; Currey, J.M.; Iwanaga, N.; He, J.; Mi, R.; et al. Endothelial cell infection and dysfunction, immune activation in severe COVID-19. Theranostics 2021, 11, 8076–8091. [Google Scholar] [CrossRef]
- Canzano, P.; Brambilla, M.; Porro, B.; Cosentino, N.; Tortorici, E.; Vicini, S.; Poggio, P.; Cascella, A.; Pengo, M.F.; Veglia, F.; et al. Platelet and Endothelial Activation as Potential Mechanisms Behind the Thrombotic Complications of COVID-19 Patients. JACC Basic Transl. Sci. 2021, 6, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Melkumyants, A.; Buryachkovskaya, L.; Lomakin, N.; Antonova, O.; Serebruany, V. Mild COVID-19 and Impaired Blood Cell-Endothelial Crosstalk: Considering Long-Term Use of Antithrombotics? Thromb. Haemost. 2021. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Rovas, A.; Osiaevi, I.; Buscher, K.; Sackarnd, J.; Tepasse, P.R.; Fobker, M.; Kuhn, J.; Braune, S.; Gobel, U.; Tholking, G.; et al. Microvascular dysfunction in COVID-19: The MYSTIC study. Angiogenesis 2021, 24, 145–157. [Google Scholar] [CrossRef]
- Stahl, K.; Gronski, P.A.; Kiyan, Y.; Seeliger, B.; Bertram, A.; Pape, T.; Welte, T.; Hoeper, M.M.; Haller, H.; David, S. Injury to the Endothelial Glycocalyx in Critically Ill Patients with COVID-19. Am. J. Respir. Crit. Care Med. 2020, 202, 1178–1181. [Google Scholar] [CrossRef]
- Lambadiari, V.; Mitrakou, A.; Kountouri, A.; Thymis, J.; Katogiannis, K.; Korakas, E.; Varlamos, C.; Andreadou, I.; Tsoumani, M.; Triantafyllidi, H.; et al. Association of COVID-19 with impaired endothelial glycocalyx, vascular function and myocardial deformation 4 months after infection. Eur. J. Heart Fail. 2021. [Google Scholar] [CrossRef]
- Fogarty, H.; Townsend, L.; Morrin, H.; Ahmad, A.; Comerford, C.; Karampini, E.; Englert, H.; Byrne, M.; Bergin, C.; O’Sullivan, J.M.; et al. Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J. Thromb. Haemost. JTH 2021, 19, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Cenko, E.; Badimon, L.; Bugiardini, R.; Claeys, M.J.; De Luca, G.; de Wit, C.; Derumeaux, G.; Dorobantu, M.; Duncker, D.J.; Eringa, E.C.; et al. Cardiovascular disease and COVID-19: A consensus paper from the ESC Working Group on Coronary Pathophysiology & Microcirculation, ESC Working Group on Thrombosis and the Association for Acute CardioVascular Care (ACVC), in collaboration with the European Heart Rhythm Association (EHRA). Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Barrett, T.J.; Cornwell, M.; Myndzar, K.; Rolling, C.C.; Xia, Y.; Drenkova, K.; Biebuyck, A.; Fields, A.T.; Tawil, M.; Luttrell-Williams, E.; et al. Platelets amplify endotheliopathy in COVID-19. Sci. Adv. 2021, 7, eabh2434. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchi, A.; Grassi, G.; Notari, S.; Gili, S.; Bordoni, V.; Tartaglia, E.; Casetti, R.; Cimini, E.; Mariotti, D.; Garotto, G.; et al. Expansion of Myeloid Derived Suppressor Cells Contributes to Platelet Activation by L-Arginine Deprivation during SARS-CoV-2 Infection. Cells 2021, 10, 2111. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, A.; Grassi, G.; Bordoni, V.; Lorenzini, P.; Cimini, E.; Casetti, R.; Tartaglia, E.; Marchioni, L.; Petrosillo, N.; Palmieri, F.; et al. Early expansion of myeloid-derived suppressor cells inhibits SARS-CoV-2 specific T-cell response and may predict fatal COVID-19 outcome. Cell Death Dis. 2020, 11, 921. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Barrett, T.J.; Bilaloglu, S.; Cornwell, M.; Burgess, H.M.; Virginio, V.W.; Drenkova, K.; Ibrahim, H.; Yuriditsky, E.; Aphinyanaphongs, Y.; Lifshitz, M.; et al. Platelets contribute to disease severity in COVID-19. J. Thromb. Haemost. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gadanec, L.K.; McSweeney, K.R.; Qaradakhi, T.; Ali, B.; Zulli, A.; Apostolopoulos, V. Can SARS-CoV-2 Virus Use Multiple Receptors to Enter Host Cells? Int. J. Mol. Sci. 2021, 22, 992. [Google Scholar] [CrossRef]
- Cunin, P.; Bouslama, R.; Machlus, K.R.; Martinez-Bonet, M.; Lee, P.Y.; Wactor, A.; Nelson-Maney, N.; Morris, A.; Guo, L.; Weyrich, A.; et al. Megakaryocyte emperipolesis mediates membrane transfer from intracytoplasmic neutrophils to platelets. eLife 2019, 8. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Petito, E.; Falcinelli, E.; Paliani, U.; Cesari, E.; Vaudo, G.; Sebastiano, M.; Cerotto, V.; Guglielmini, G.; Gori, F.; Malvestiti, M.; et al. Association of Neutrophil Activation, More Than Platelet Activation, With Thrombotic Complications in Coronavirus Disease 2019. J. Infect. Dis. 2021, 223, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Comer, S.P.; Cullivan, S.; Szklanna, P.B.; Weiss, L.; Cullen, S.; Kelliher, S.; Smolenski, A.; Murphy, C.; Altaie, H.; Curran, J.; et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol. 2021, 19, e3001109. [Google Scholar] [CrossRef]
- Cappellano, G.; Raineri, D.; Rolla, R.; Giordano, M.; Puricelli, C.; Vilardo, B.; Manfredi, M.; Cantaluppi, V.; Sainaghi, P.P.; Castello, L.; et al. Circulating Platelet-Derived Extracellular Vesicles Are a Hallmark of Sars-Cov-2 Infection. Cells 2021, 10, 85. [Google Scholar] [CrossRef]
- Fricke, A.; Ullrich, P.V.; Cimniak, A.F.V.; Becherer, C.; Follo, M.; Heinz, J.; Scholber, J.; Herget, G.W.; Hauschild, O.; Wittel, U.A.; et al. Levels of activated platelet-derived microvesicles in patients with soft tissue sarcoma correlate with an increased risk of venous thromboembolism. BMC Cancer 2017, 17, 527. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Back, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Fiolet, A.T.L.; Opstal, T.S.J.; Mosterd, A.; Eikelboom, J.W.; Jolly, S.S.; Keech, A.C.; Kelly, P.; Tong, D.C.; Layland, J.; Nidorf, S.M.; et al. Efficacy and safety of low-dose colchicine in patients with coronary disease: A systematic review and meta-analysis of randomized trials. Eur. Heart J. 2021, 42, 2765–2775. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Ljung, L.; Rantapaa-Dahlqvist, S.; Jacobsson, L.T.; Askling, J. Response to biological treatment and subsequent risk of coronary events in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Padfield, G.J.; Din, J.N.; Koushiappi, E.; Mills, N.L.; Robinson, S.D.; Cruden Nle, M.; Lucking, A.J.; Chia, S.; Harding, S.A.; Newby, D.E. Cardiovascular effects of tumour necrosis factor alpha antagonism in patients with acute myocardial infarction: A first in human study. Heart 2013, 99, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.B.; Nielsen, C.; Nybo, M.; Just, S.A.; Vinholt, P.J. The in vitro effect of antirheumatic drugs on platelet function. Platelets 2020, 31, 248–257. [Google Scholar] [CrossRef]
- Shah, B.; Allen, N.; Harchandani, B.; Pillinger, M.; Katz, S.; Sedlis, S.P.; Echagarruga, C.; Samuels, S.K.; Morina, P.; Singh, P.; et al. Effect of Colchicine on Platelet-Platelet and Platelet-Leukocyte Interactions: A Pilot Study in Healthy Subjects. Inflammation 2016, 39, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nisio, M.; Potere, N.; Candeloro, M.; Spacone, A.; Pieramati, L.; Ferrandu, G.; Rizzo, G.; La Vella, M.; Di Carlo, S.; Cibelli, D.; et al. Interleukin-6 receptor blockade with subcutaneous tocilizumab improves coagulation activity in patients with COVID-19. Eur. J. Intern. Med. 2021, 83, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Gergi, M.; Cushman, M.; Littenberg, B.; Budd, R.C. Thrombo-inflammation response to Tocilizumab in COVID-19. Res. Pract. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, P.; Taglialatela, V.; Pellegrino, G.; Morello, A.; Conte, S.; Di Serafino, L.; Cimmino, G. Effects of colchicine on platelet aggregation in patients on dual antiplatelet therapy with aspirin and clopidogrel. J. Thromb. Thrombolysis 2020, 50, 468–472. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, G.; Tarallo, R.; Conte, S.; Morello, A.; Pellegrino, G.; Loffredo, F.S.; Cali, G.; De Luca, N.; Golino, P.; Trimarco, B.; et al. Colchicine reduces platelet aggregation by modulating cytoskeleton rearrangement via inhibition of cofilin and LIM domain kinase 1. Vasc. Pharmacol. 2018, 111, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Pennings, G.J.; Reddel, C.J.; Traini, M.; Campbell, H.; Chen, V.; Kritharides, L. Colchicine inhibits ROS generation in response to glycoprotein VI stimulation. Sci. Rep. 2021, 11, 11965. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Santilli, F.; Guerci, M.; Simeone, P.; Ardizzone, S.; Massari, A.; Giuffrida, P.; Tripaldi, R.; Malara, A.; Liani, R.; et al. Oxidative stress and thromboxane-dependent platelet activation in inflammatory bowel disease: Effects of anti-TNF-alpha treatment. Thromb. Haemost. 2016, 116, 486–495. [Google Scholar] [CrossRef]
- Menchen, L.; Marin-Jimenez, I.; Arias-Salgado, E.G.; Fontela, T.; Hernandez-Sampelayo, P.; Rodriguez, M.C.; Butta, N.V. Matrix metalloproteinase 9 is involved in Crohn’s disease-associated platelet hyperactivation through the release of soluble CD40 ligand. Gut 2009, 58, 920–928. [Google Scholar] [CrossRef]
- DeSena, A.D.; Do, T.; Schulert, G.S. Systemic autoinflammation with intractable epilepsy managed with interleukin-1 blockade. J. Neuroinflamm. 2018, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Wang, C.; Liu, Y.; Li, B.; Zhang, W.; Wang, L.; Yu, M.; Zhao, X.; Du, J.; Zhang, J.; et al. Neutrophil Extracellular Traps Induce Intestinal Damage and Thrombotic Tendency in Inflammatory Bowel Disease. J. Crohn’s Colitis 2020, 14, 240–253. [Google Scholar] [CrossRef]
- Carminita, E.; Crescence, L.; Brouilly, N.; Altie, A.; Panicot-Dubois, L.; Dubois, C. DNAse-dependent, NET-independent pathway of thrombus formation in vivo. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Okur, H.K.; Yalcin, K.; Tastan, C.; Demir, S.; Yurtsever, B.; Karakus, G.S.; Kancagi, D.D.; Abanuz, S.; Seyis, U.; Zengin, R.; et al. Preliminary report of in vitro and in vivo effectiveness of dornase alfa on SARS-CoV-2 infection. New Microbes New Infect. 2020, 37, 100756. [Google Scholar] [CrossRef]
- Weber, A.G.; Chau, A.S.; Egeblad, M.; Barnes, B.J.; Janowitz, T. Nebulized in-line endotracheal dornase alfa and albuterol administered to mechanically ventilated COVID-19 patients: A case series. Mol. Med. 2020, 26, 91. [Google Scholar] [CrossRef] [PubMed]
- Toma, A.; Darwish, C.; Taylor, M.; Harlacher, J.; Darwish, R. The Use of Dornase Alfa in the Management of COVID-19-Associated Adult Respiratory Distress Syndrome. Crit. Care Res. Pract. 2021, 2021, 8881115. [Google Scholar] [CrossRef]
- Novotny, J.; Chandraratne, S.; Weinberger, T.; Philippi, V.; Stark, K.; Ehrlich, A.; Pircher, J.; Konrad, I.; Oberdieck, P.; Titova, A.; et al. Histological comparison of arterial thrombi in mice and men and the influence of Cl-amidine on thrombus formation. PLoS ONE 2018, 13, e0190728. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, M.; Chen, K.; Chen, B.; Zhao, Y.; Gong, H.; Zhao, X.; Qi, R. Suppression of TLR4 activation by resveratrol is associated with STAT3 and Akt inhibition in oxidized low-density lipoprotein-activated platelets. Eur. J. Pharmacol. 2018, 836, 1–10. [Google Scholar] [CrossRef]
- Hally, K.E.; La Flamme, A.C.; Harding, S.A.; Larsen, P.D. The effects of aspirin and ticagrelor on Toll-like receptor (TLR)-mediated platelet activation: Results of a randomized, cross-over trial. Platelets 2019, 30, 599–607. [Google Scholar] [CrossRef]
- Kovacevic, K.D.; Buchtele, N.; Schoergenhofer, C.; Derhaschnig, U.; Gelbenegger, G.; Brostjan, C.; Zhu, S.; Gilbert, J.C.; Jilma, B. The aptamer BT200 effectively inhibits von Willebrand factor (VWF) dependent platelet function after stimulated VWF release by desmopressin or endotoxin. Sci. Rep. 2020, 10, 11180. [Google Scholar] [CrossRef]
- Nimjee, S.M.; Dornbos, D., 3rd; Pitoc, G.A.; Wheeler, D.G.; Layzer, J.M.; Venetos, N.; Huttinger, A.; Talentino, S.E.; Musgrave, N.J.; Moody, H.; et al. Preclinical Development of a vWF Aptamer to Limit Thrombosis and Engender Arterial Recanalization of Occluded Vessels. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1228–1241. [Google Scholar] [CrossRef]
- Nicolson, P.L.R.; Nock, S.H.; Hinds, J.; Garcia-Quintanilla, L.; Smith, C.W.; Campos, J.; Brill, A.; Pike, J.A.; Khan, A.O.; Poulter, N.S.; et al. Low-dose Btk inhibitors selectively block platelet activation by CLEC-2. Haematologica 2021, 106, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobie, G.; Kuriri, F.A.; Omar, M.M.A.; Alanazi, F.; Gazwani, A.M.; Tang, C.P.S.; Sze, D.M.; Handunnetti, S.M.; Tam, C.; Jackson, D.E. Ibrutinib, but not zanubrutinib, induces platelet receptor shedding of GPIb-IX-V complex and integrin alphaIIbbeta3 in mice and humans. Blood Adv. 2019, 3, 4298–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninomoto, J.; Mokatrin, A.; Kinoshita, T.; Marimpietri, C.; Barrett, T.D.; Chang, B.Y.; Sukbuntherng, J.; James, D.F.; Crowther, M. Effects of ibrutinib on in vitro platelet aggregation in blood samples from healthy donors and donors with platelet dysfunction. Hematology 2020, 25, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelius, D.C.; Travis, O.K.; Tramel, R.W.; Borges-Rodriguez, M.; Baik, C.H.; Greer, M.; Giachelli, C.A.; Tardo, G.A.; Williams, J.M. NLRP3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS ONE 2020, 15, e0234039. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | Agent | Population | Findings |
|---|---|---|---|
| Di Sabatino et al. [165] | Infliximab | IBD | ↓ TX biosynthesis ↓ sCD40L |
| Manfredi et al. [86] | Anti-TNF-α | RA | ↓ platelet-leukocyte and platelet-monocyte aggregates ↓ P-selectin |
| Menchen et al. [166] | Infliximab | CD | ↓ sCD40L Non-significant reduction of the enhanced binding of FITC–fibrinogen |
| Padfield et al. [157] | Etanercept | AMI | Non-significant effects on platelet activation parameters |
| Nielsen et al. [158] | Adalimumab | In vitro | ↑ TRAP-induced platelet aggregation ≥20% |
| Tocilizumab | ↓ ADP- and collagen-induced platelet aggregation ≥20% | ||
| Canzano et al. [128] | Tocilizumab | COVID-19 (in vitro) | ↓ P-Selectin ↓ Platelet-granulocyte and platelet-neutrophil aggregates |
| Shah et al. [159] | Colchicine | Healthy | ↓ P-Selectin and PAC-1 ↓ Platelet-granulocyte and platelet-neutrophil aggregates No effect on homotypic platelet aggregation |
| Cirillo et al. [162] | Colchicine | CAD | ↓ TRAP-induced platelet aggregation |
| Cimmino [163] | Colchicine | CAD (in vitro) | ADP/Collagen/TRAP-induced platelet aggregation due to cytoskeleton reorganization |
| Pennings [164] | Colchicine | Healthy (in vitro) | ↓ P-Selectin ↓ collagen-induced platelet aggregation |
| DeSena et al. [167] | Canakinumab | Case of epilepsy | Downregulation of genes associated with platelet activation |
| Study | Agent | Target | Findings |
|---|---|---|---|
| Li et al. [168] | DNAse | NETs | ↓ platelet activation ↓ thrombus formation |
| Carminita et al. [169] | DNAse | NETs | ↓ ATP and ADP-induced thrombus formation |
| Novotny et al. [173] | Chloramidine | NETs | ↓ arterial thrombus formation |
| Clark et al. [174] | Eritoran | TLR4 | ↓ platelet-leukocyte aggregation ↓ platelet-mediated neutrophil degranulation |
| Sun et al. [175] | Resveratrol | TLR4 | ↓ TRAP- and collagen-induced platelet aggregation ↓ platelet adhesion and secretion ↓CD40L and platelet factor-4 |
| Kovacevic et al. [177] | BT200 | vWF | Inhibition of high shear rate- and ristocetin-induced platelet aggregation |
| Nimjee et al. [178] | DTRI-031 | vWF | Inhibition of platelet aggregation and thrombosis |
| Dobie et al. [180] | Ibrutinib | BTK | ADAM17-mediated shedding of GPIbα and GPIX in vitro and of GPIbα in vivo |
| Ninomoto et al. [181] | Ibrutinib | BTK | ↓ collagen-induced platelet aggregation |
| Nicolson et al. [179] | Ibrutinib | BTK | ↓ rhodocytin-induced platelet aggregation and granule secretion |
| Acalabrutinib | ↓ rhodocytin- and podoplanin-induced platelet aggregation Blocks CLEC-2-mediated platelet activation and granule secretion | ||
| Cornelius et al. [182] | MCC950 | NLRP3 inflammasome | ↓ platelet activation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theofilis, P.; Sagris, M.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19. Int. J. Mol. Sci. 2021, 22, 11170. https://doi.org/10.3390/ijms222011170
Theofilis P, Sagris M, Antonopoulos AS, Oikonomou E, Tsioufis C, Tousoulis D. Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19. International Journal of Molecular Sciences. 2021; 22(20):11170. https://doi.org/10.3390/ijms222011170
Chicago/Turabian StyleTheofilis, Panagiotis, Marios Sagris, Alexios S. Antonopoulos, Evangelos Oikonomou, Costas Tsioufis, and Dimitris Tousoulis. 2021. "Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19" International Journal of Molecular Sciences 22, no. 20: 11170. https://doi.org/10.3390/ijms222011170