
Discovery of Pyrano[2,3-c]pyrazole Derivatives as Novel Potential Human Coronavirus Inhibitors: Design, Synthesis, In Silico, In Vitro, and ADME Studies
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
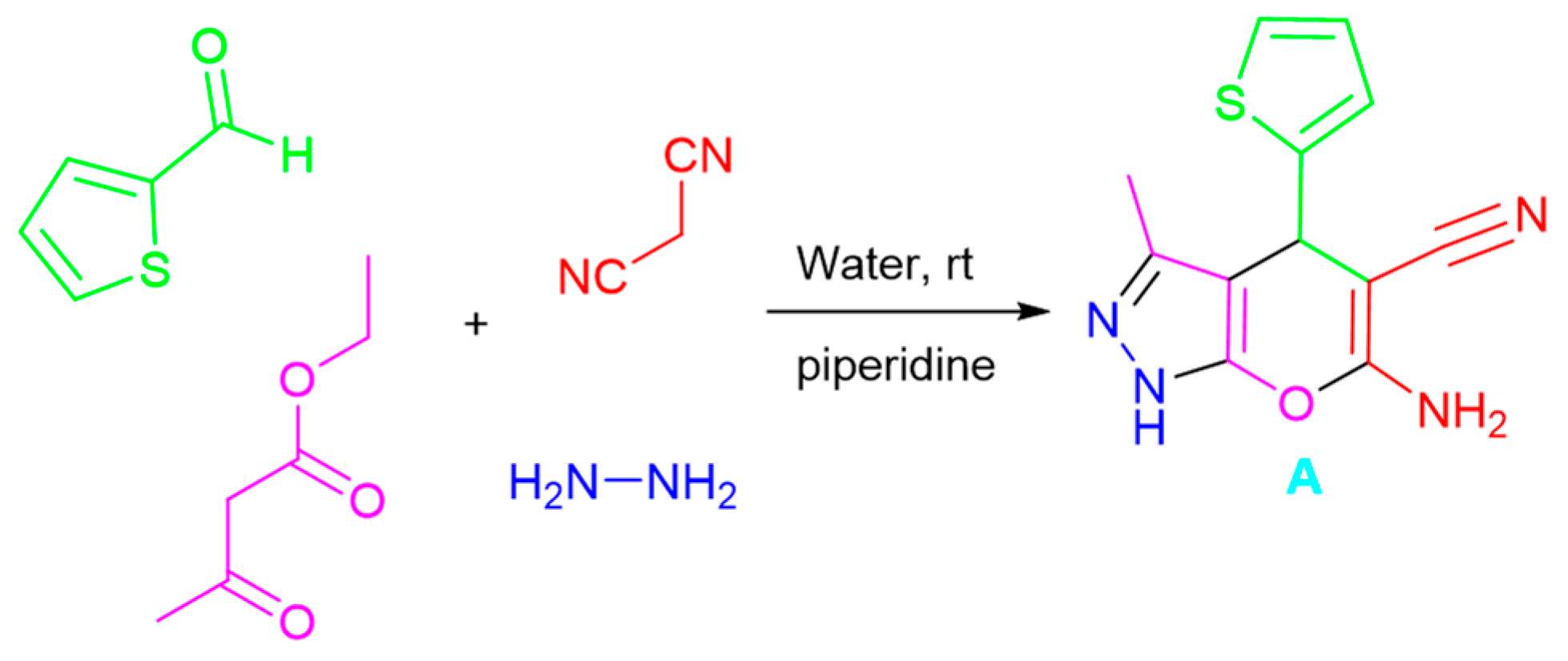
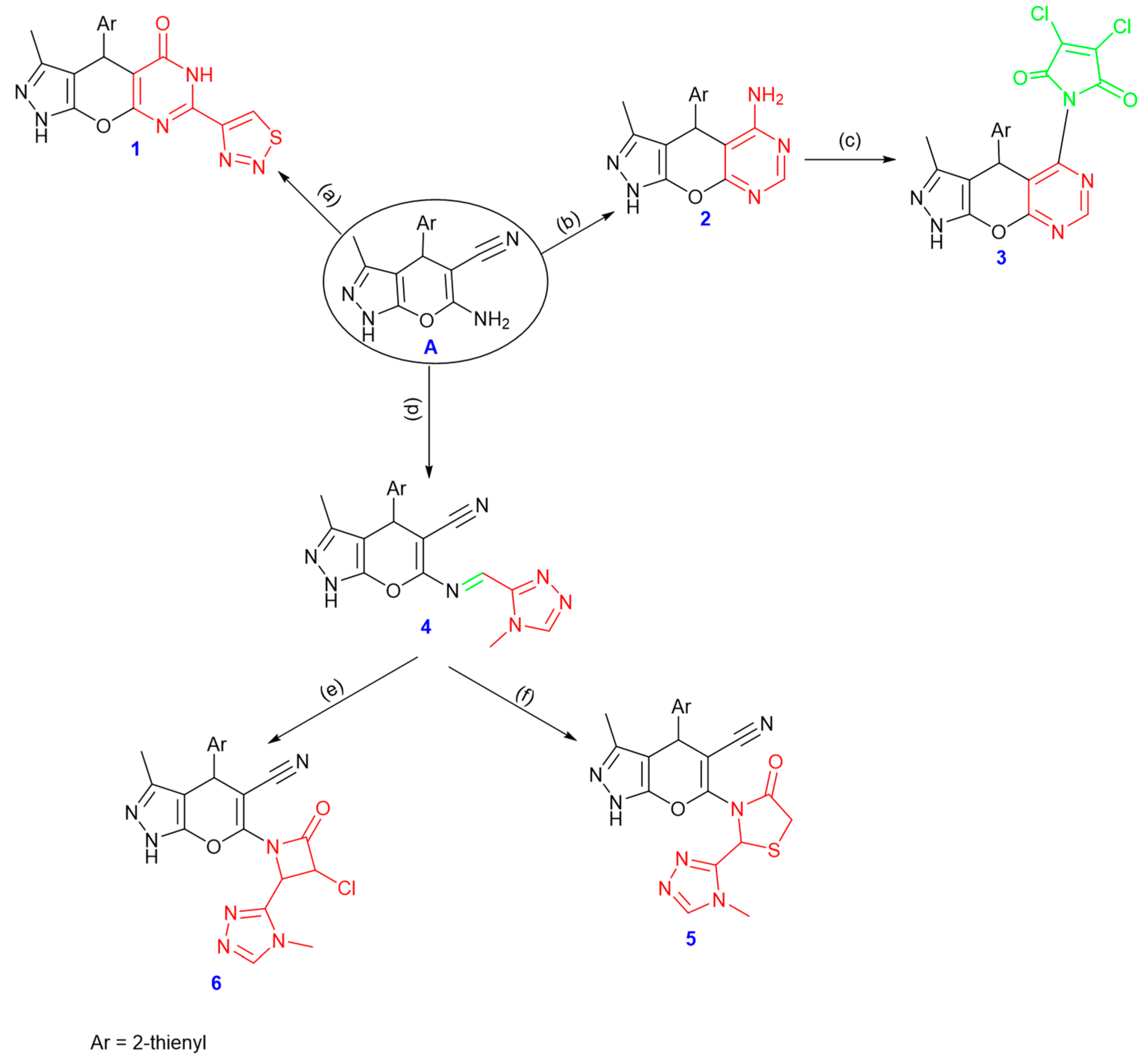
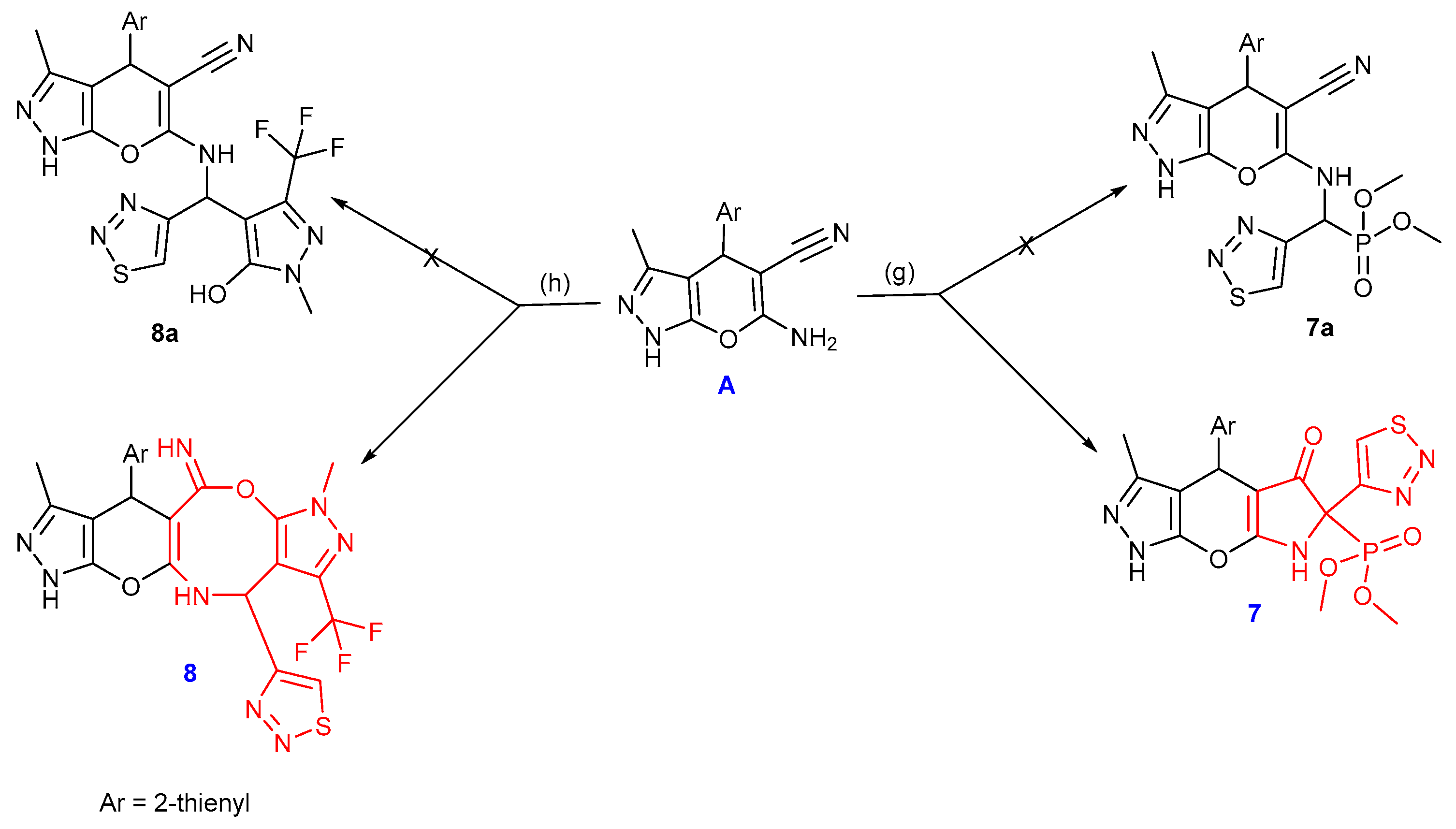
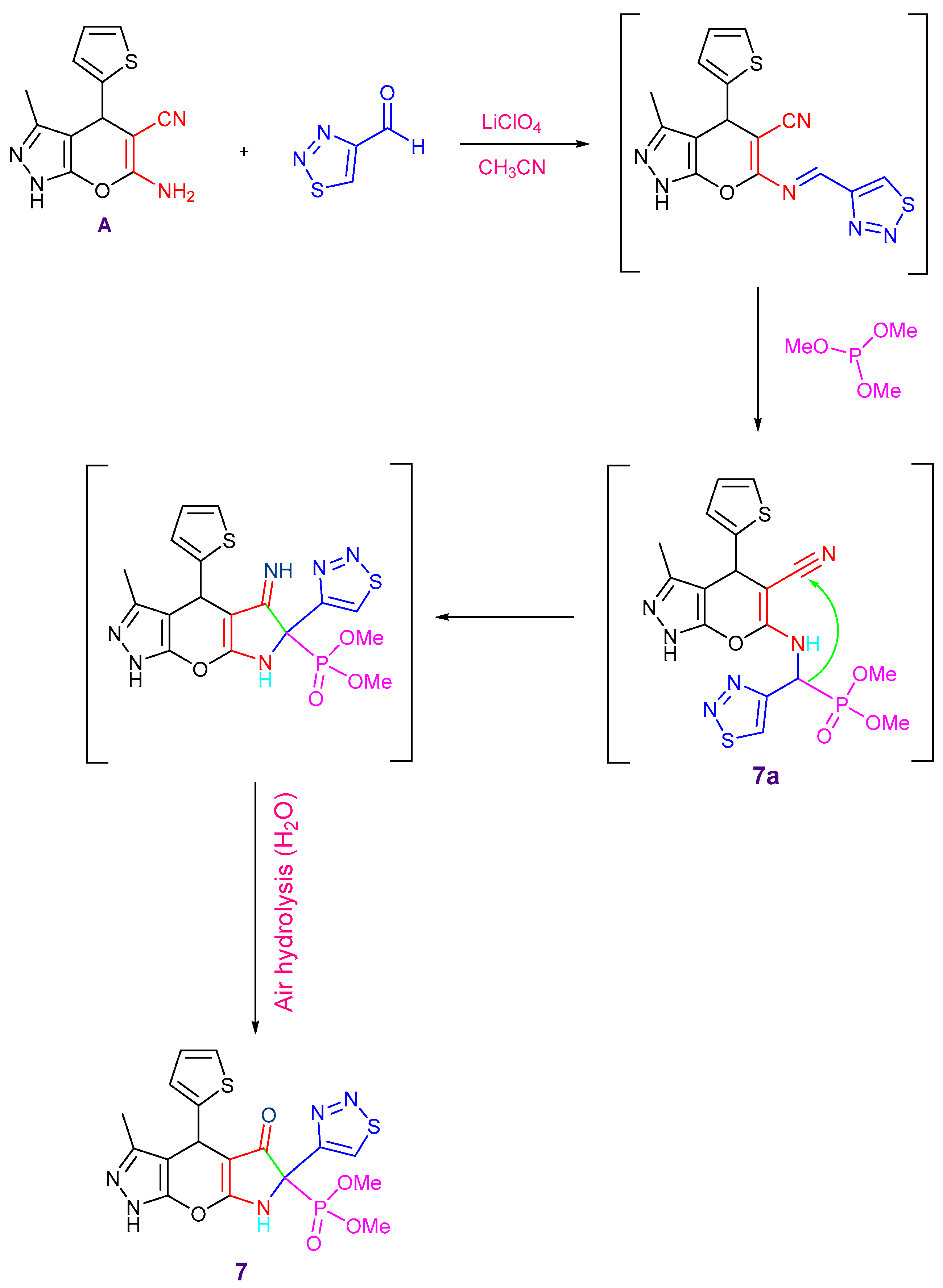
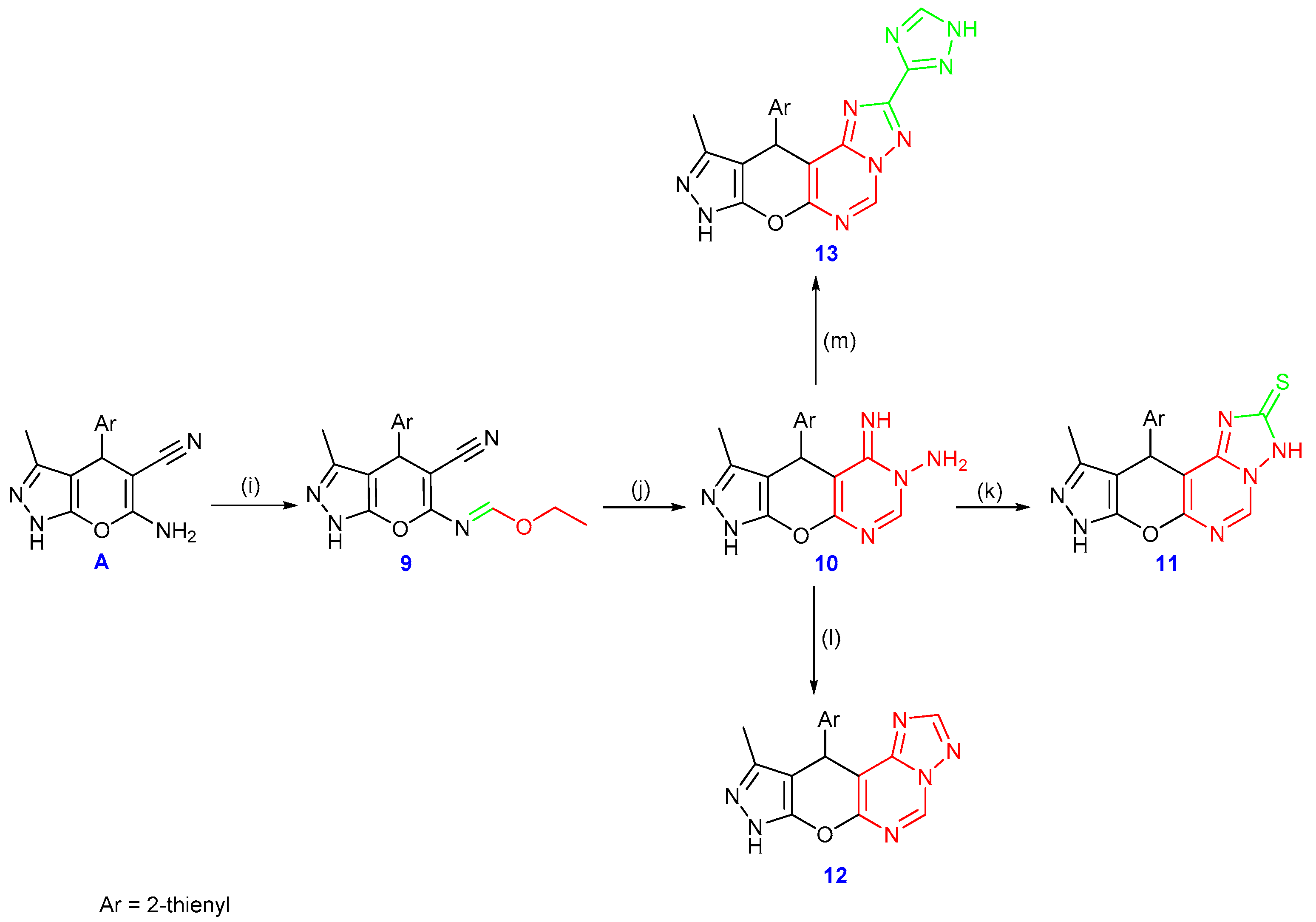
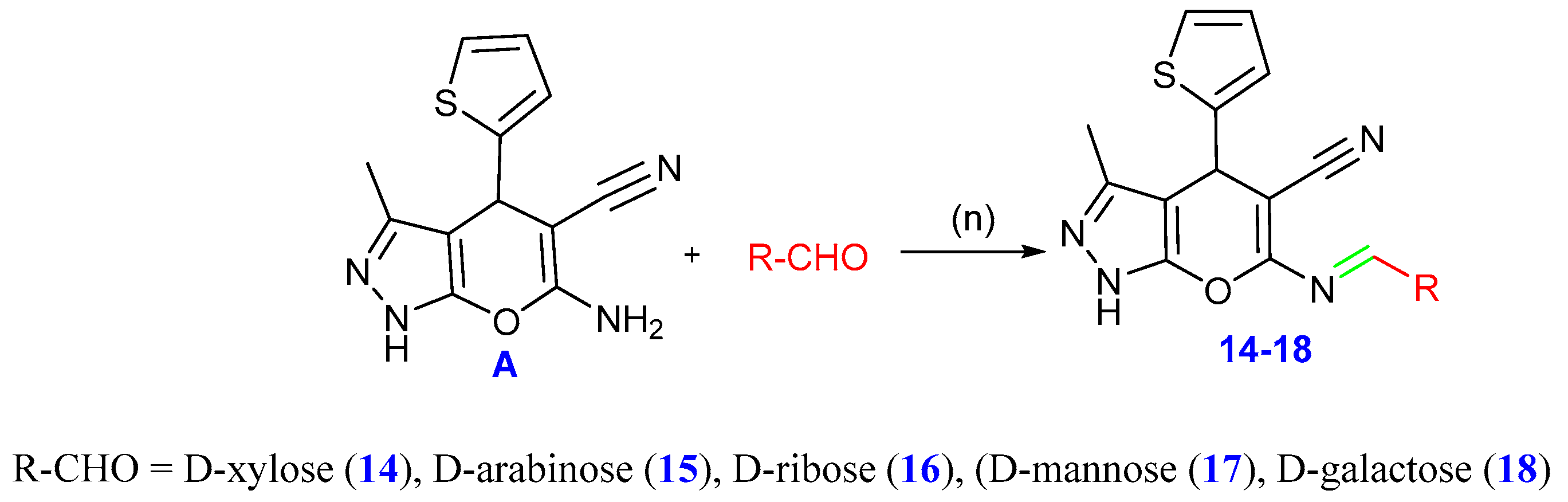
2.1. Chemistry
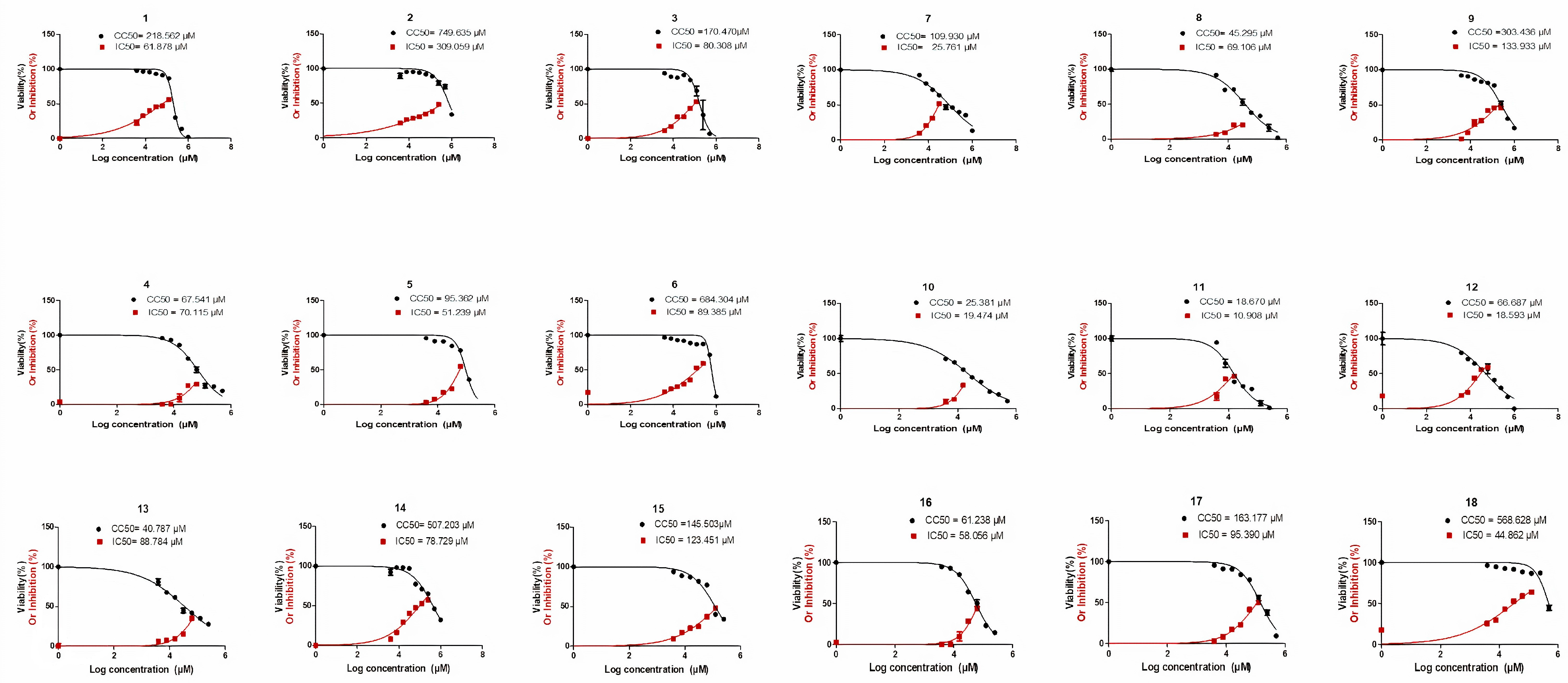
2.2. Biological Evaluation
2.3. Computational Studies
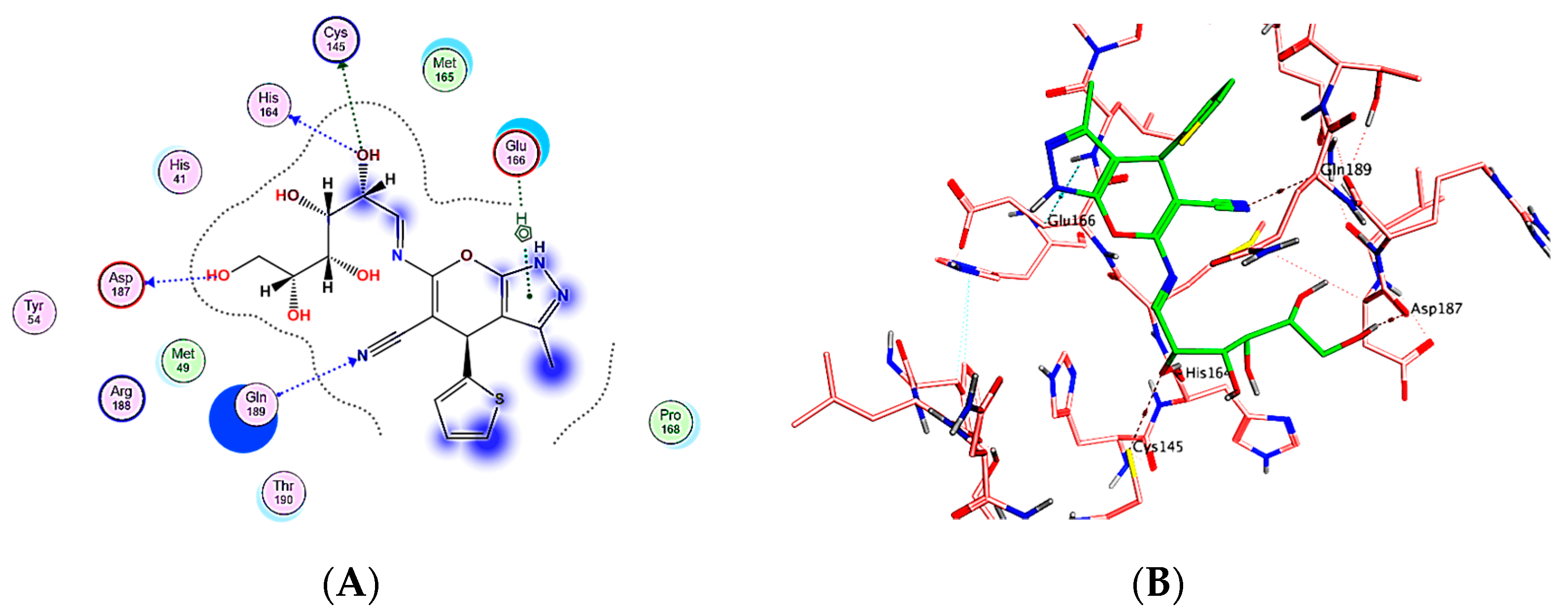
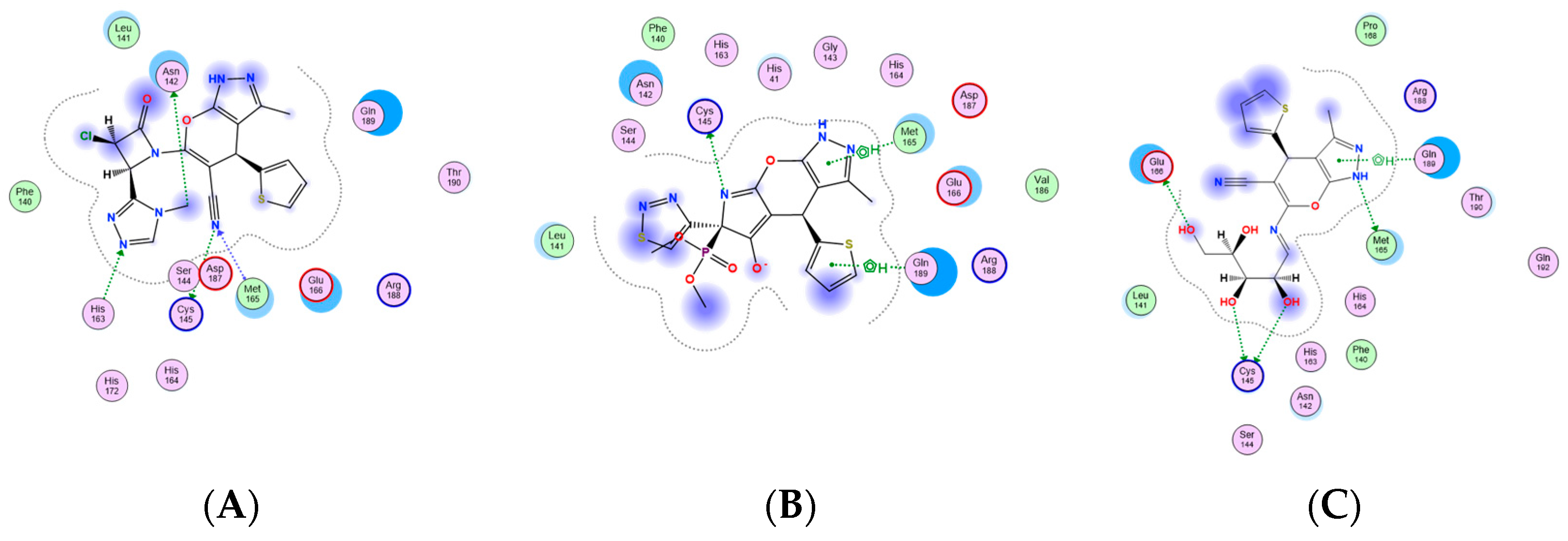
2.3.1. Docking Simulations
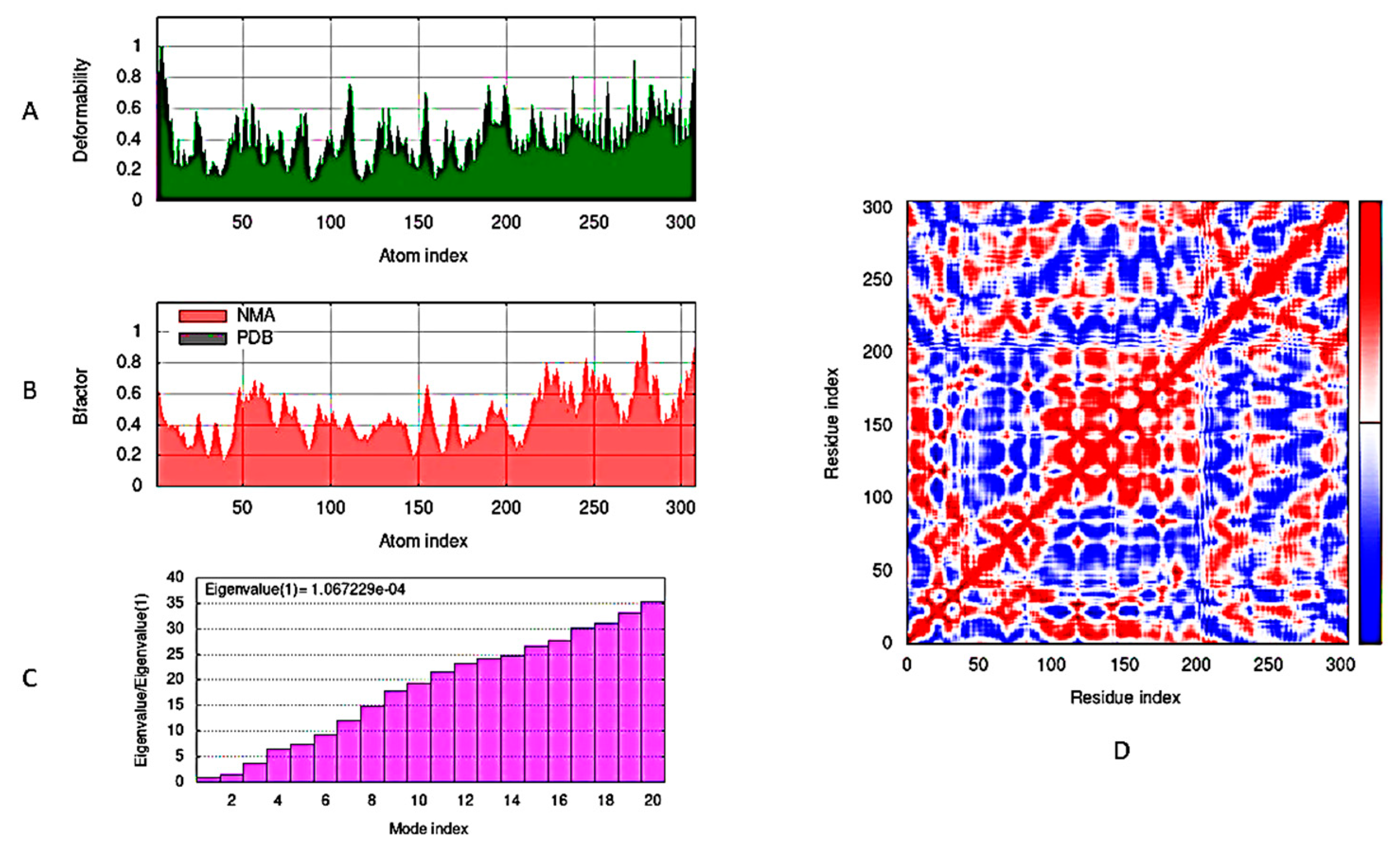
2.3.2. Molecular Dynamic Simulations
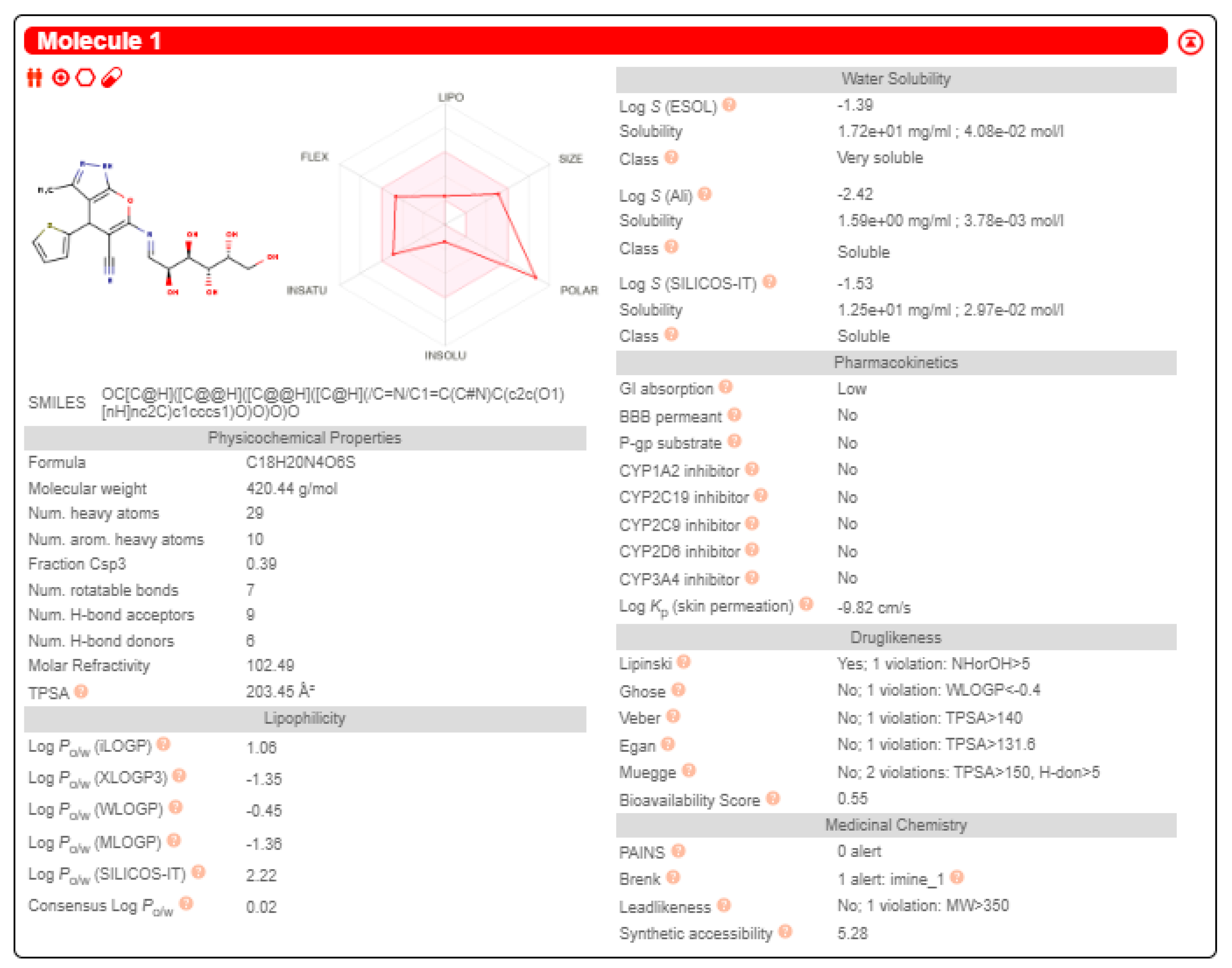
2.3.3. Swiss-ADME Study
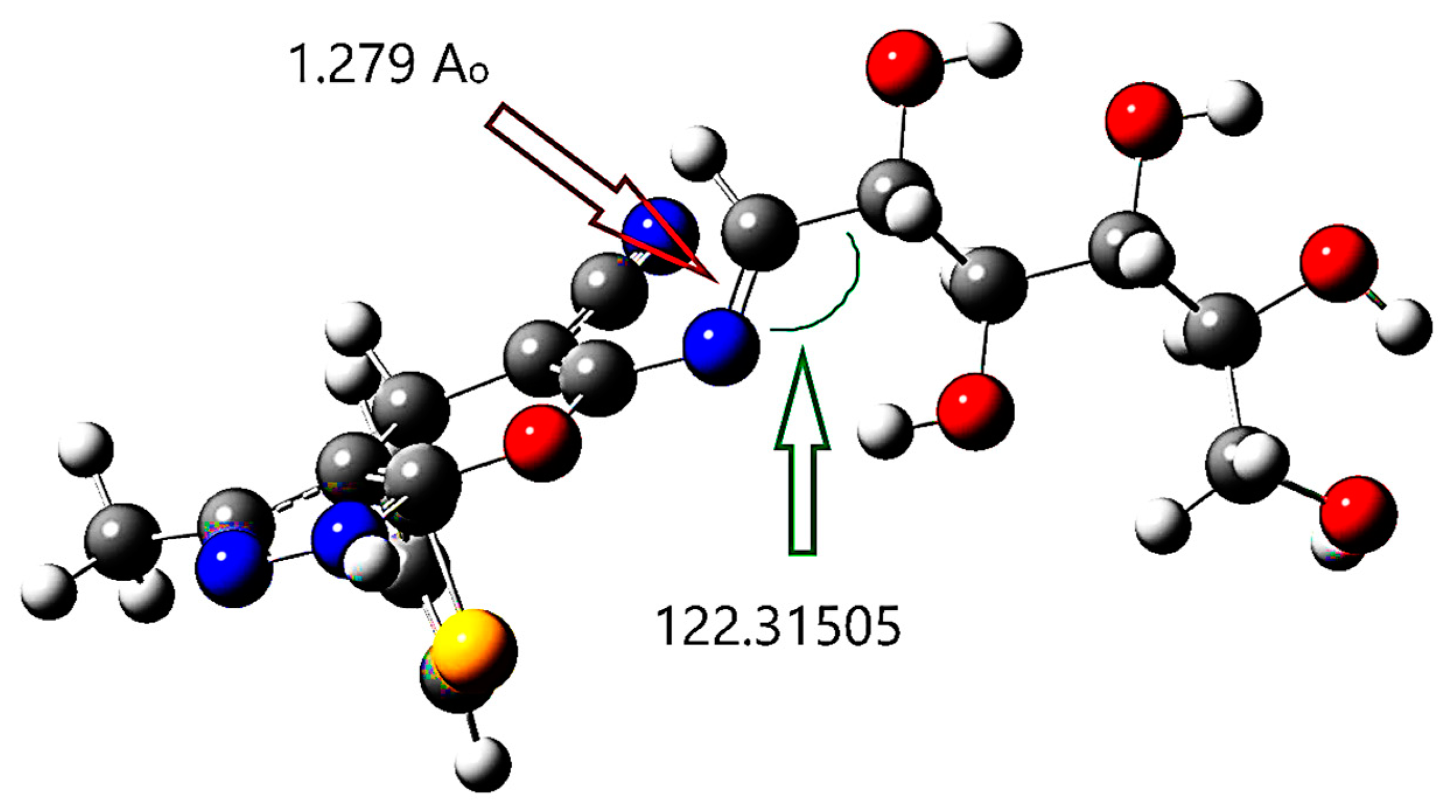
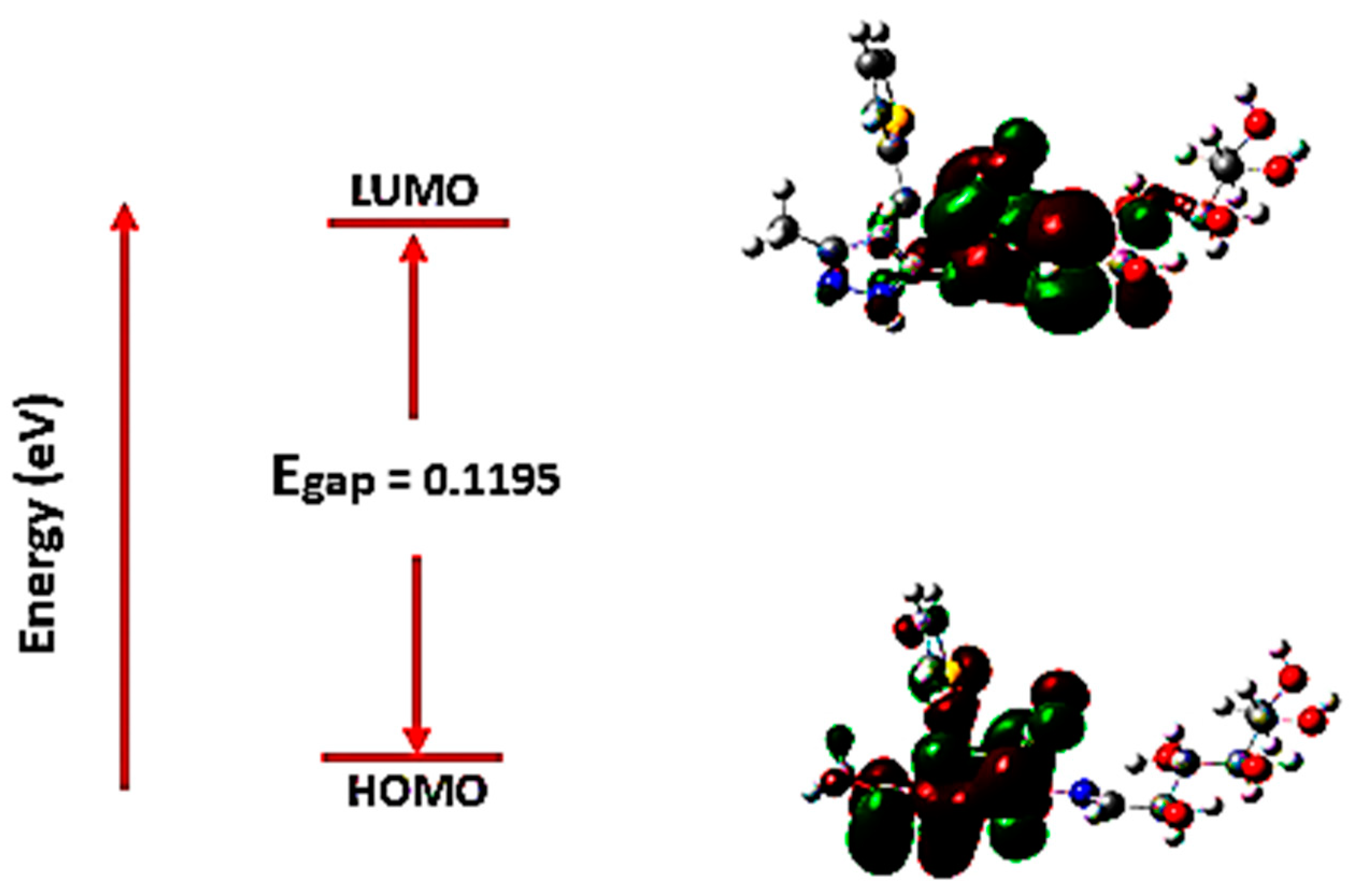
2.3.4. Density Function Theory (DFT)
Structure Optimization
Frontier Molecular Orbital Analysis
3. Materials and Methods
3.1. Chemistry
3.2. Biological Assay
3.2.1. Viruses and Cells
3.2.2. MTT Assay for Cellular Toxicity
3.2.3. Cytopathic Inhibition Assay
3.2.4. Calculating IC50, CC50, and SI Values
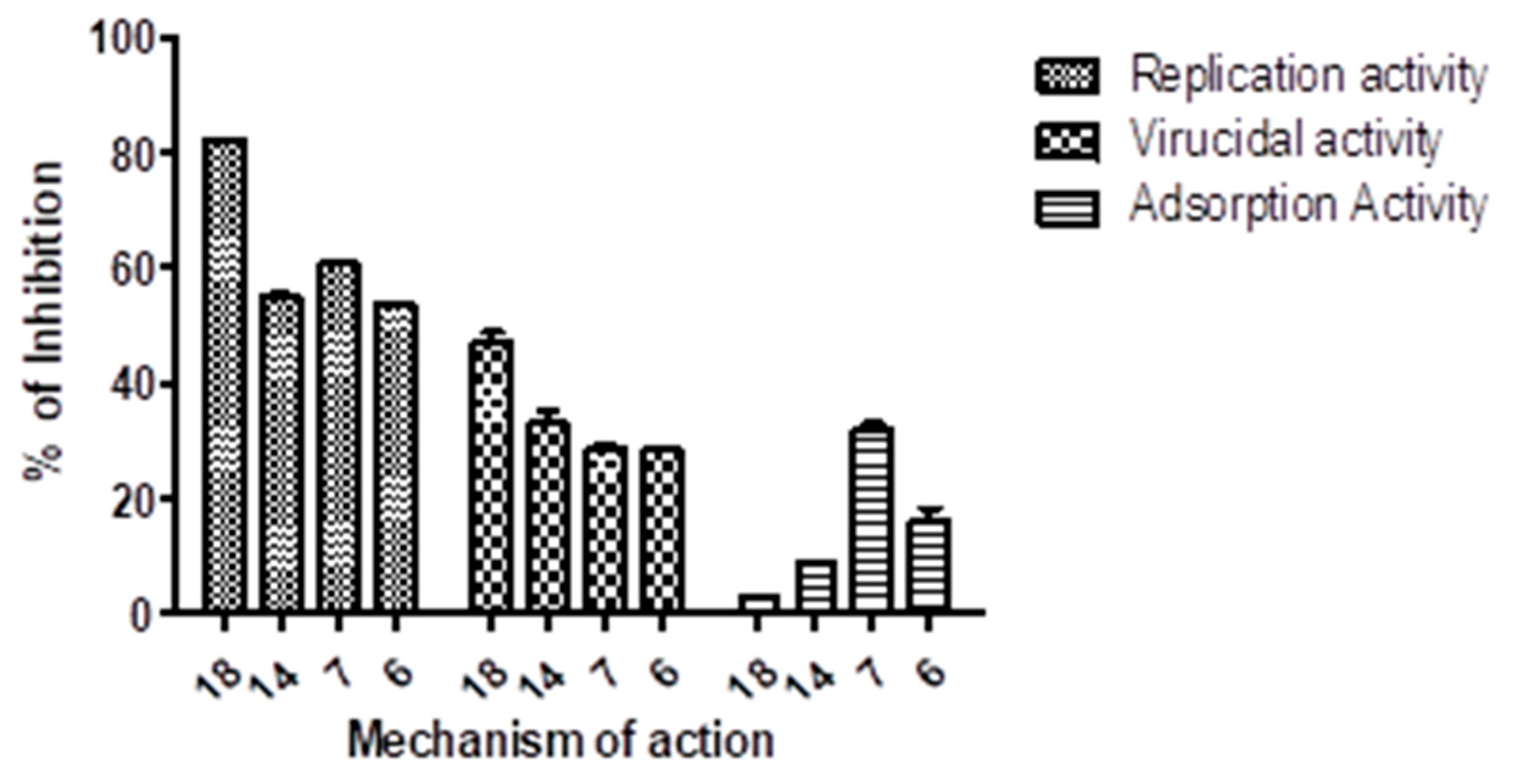
3.2.5. Mode of Action Using Plaque Assay
3.2.6. Virucidal Mechanism
3.2.7. Adsorption Mechanism
3.2.8. Replication Mechanism
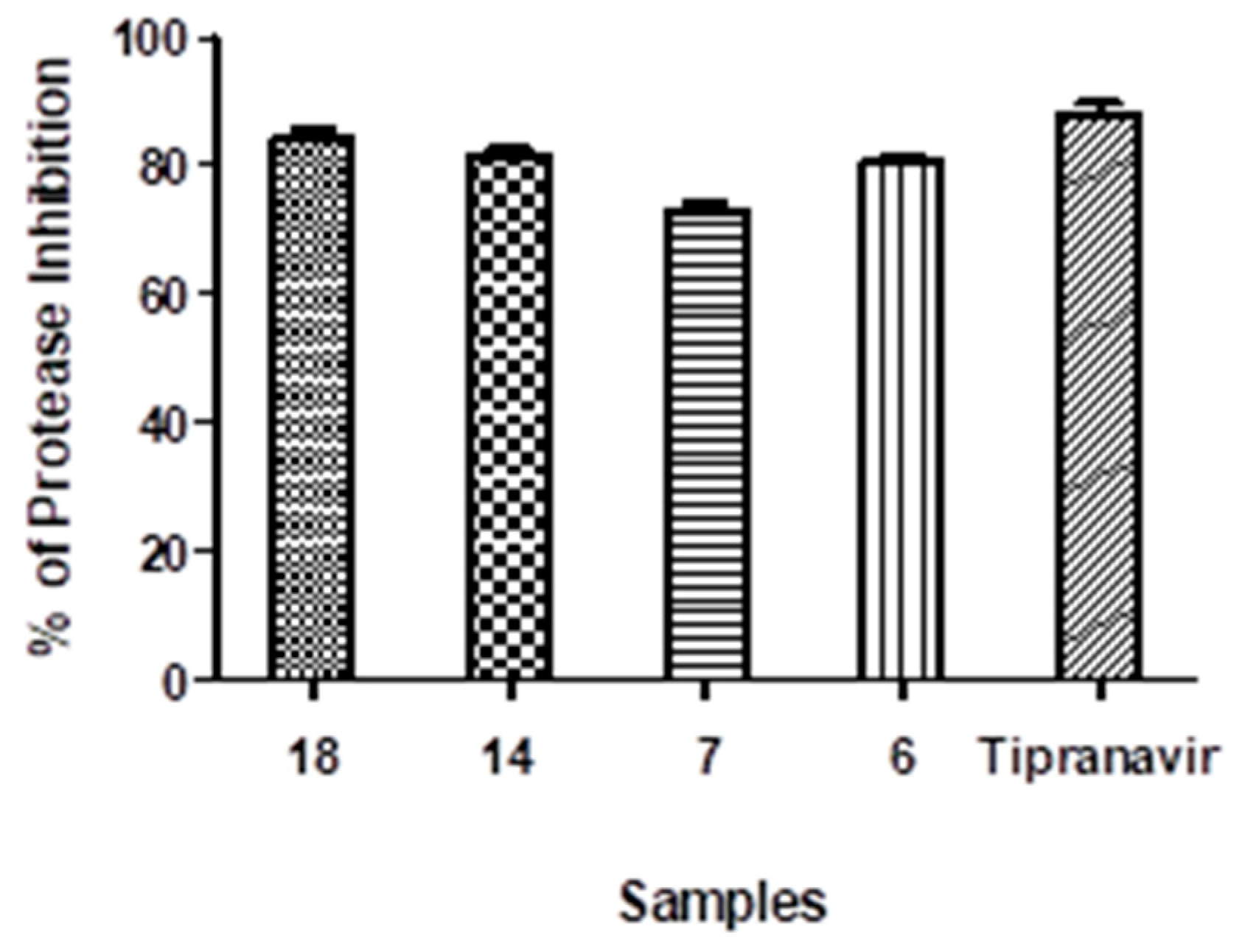
3.2.9. Protease (SARS-CoV2) Inhibition Test
3.2.10. Statistical Investigation
3.3. Computational Investigations
3.3.1. Docking Simulations
3.3.2. The Target Protein Structure Preparation
3.3.3. The Investigated Drug Molecules’ Preparation
3.3.4. Docking Validation
3.3.5. Molecular Dynamic Simulations
3.3.6. Swiss-ADME Study
3.3.7. Density Function Theory (DFT)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alamshany, Z.M.; Khattab, R.R.; Hassan, N.A.; El-Sayed, A.A.; Tantawy, M.A.; Mostafa, A.; Hassan, A.A. Synthesis and Molecular Docking Study of Novel Pyrimidine Derivatives against COVID-19. Molecules 2023, 28, 739. [Google Scholar] [CrossRef]
- Chandel, V.; Raj, S.; Rathi, B.; Kumar, D. In silico identification of potent FDA approved drugs against Coronavirus COVID-19 main protease: A drug repurposing approach In silico identification of potent FDA approved drugs against Coronavirus COVID-19 main protease: A drug repurposing approach. Chem. Biol. Lett. 2020, 7, 166–175. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sharma, A.; Nandi, S.P. Identification of Potent Inhibitors of COVID-19 Main Protease Enzyme by Molecular Docking Study. ChemXiv 2020, 1, 1–10. [Google Scholar] [CrossRef]
- Addoum, B.; El Khalfi, B.; Sakoui, S.; Derdak, R.; Elmakssoudi, A.; Soukri, A. Synthesis and molecular docking studies of some pyrano [2,3-c] pyrazole as an Inhibitor of SARS-Coronavirus 3CL protease. Lett. Appl. NanoBiosci. 2022, 11, 3780–3801. [Google Scholar] [CrossRef]
- Adil, M.A.; Rahman, R.; Whitelaw, D.; Jain, V.; Al-Taan, O.; Rashid, F.; Munasinghe, A.; Jambulingam, P. SARS-CoV-2 and the pandemic of COVID-19. Postgrad. Med. J. 2021, 97, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Grellet, E.; Goulet, A.; Imbert, I. Replication of the coronavirus genome: A paradox among positive-strand RNA viruses. J. Biol. Chem. 2022, 298, 101923. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Tan, H.; Hu, Y.; Jadhav, P.; Tan, B.; Wang, J. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. J. Med. Chem. 2022, 65, 7561–7580. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: State of the art and future opportunities. J. Med. Chem. 2022, 65, 2716–2746. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of COVID-19–final report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Chera, A.; Tanca, A. Remdesivir: The first FDA-approved anti-COVID-19 treatment for young children. Discoveries 2022, 10, 151. [Google Scholar] [CrossRef]
- Tanne, J.H. COVID-19: FDA authorizes pharmacists to prescribe Paxlovid. BMJ 2022, 378, 1695. [Google Scholar] [CrossRef]
- Hersi, F.; Sebastian, A.; Tarazi, H.; Srinivasulu, V.; Mostafa, A.; Allayeh, A.K.; Zeng, C.; Hachim, I.Y.; Liu, S.L.; Abu-Yousef, I.A.; et al. Discovery of novel papain-like protease inhibitors for potential treatment of COVID-19. Eur. J. Med. Chem. 2023, 254, 115380. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzova, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Hobartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Menendez, J.C. Approaches to the potential therapy of COVID-19: A general overview from the medicinal chemistry perspective. Molecules 2022, 27, 658. [Google Scholar] [CrossRef]
- Mohamed, Y.; El-Maradny, Y.A.; Saleh, A.K.; Nayl, A.A.; El-Gendi, H.; El-Fakharany, E.M. A comprehensive insight into current control of COVID-19: Immunogenicity, vaccination, and treatment. Biomed. Pharmacother. 2022, 153, 113499. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef]
- Ton, A.T.; Pandey, M.; Smith, J.R.; Ban, F.; Fernandez, M.; Cherkasov, A. Targeting SARS-CoV-2 papain-like protease in the postvaccine era. Trends Pharmacol. Sci. 2022, 43, 906–919. [Google Scholar] [CrossRef]
- Malebari, A.M.; Ahmed, H.E.; Ihmaid, S.K.; Omar, A.M.; Muhammad, Y.A.; Althagfan, S.S.; Aljuhani, N.; El-Sayed, A.A.A.; Halawa, A.H.; El-Tahir, H.M.; et al. Exploring the dual effect of novel 1,4-diarylpyranopyrazoles as antiviral and anti-inflammatory for the management of SARS-CoV-2 and associated inflammatory symptoms. Bioorg. Chem. 2023, 130, 106255. [Google Scholar] [CrossRef]
- da Silva, F.M.A.; da Silva, K.P.A.; de Oliveira, L.P.M.; Costa, E.V.; Koolen, H.H.; Pinheiro, M.L.B.; de Souza, A.Q.L.; de Souza, A.D.L. Flavonoid glycosides and their putative human metabolites as potential inhibitors of the SARS-CoV-2 main protease (Mpro) and RNA-dependent RNA polymerase (RdRp). Mem. Inst. Oswaldo Cruz. 2020, 115, e200207. [Google Scholar] [CrossRef]
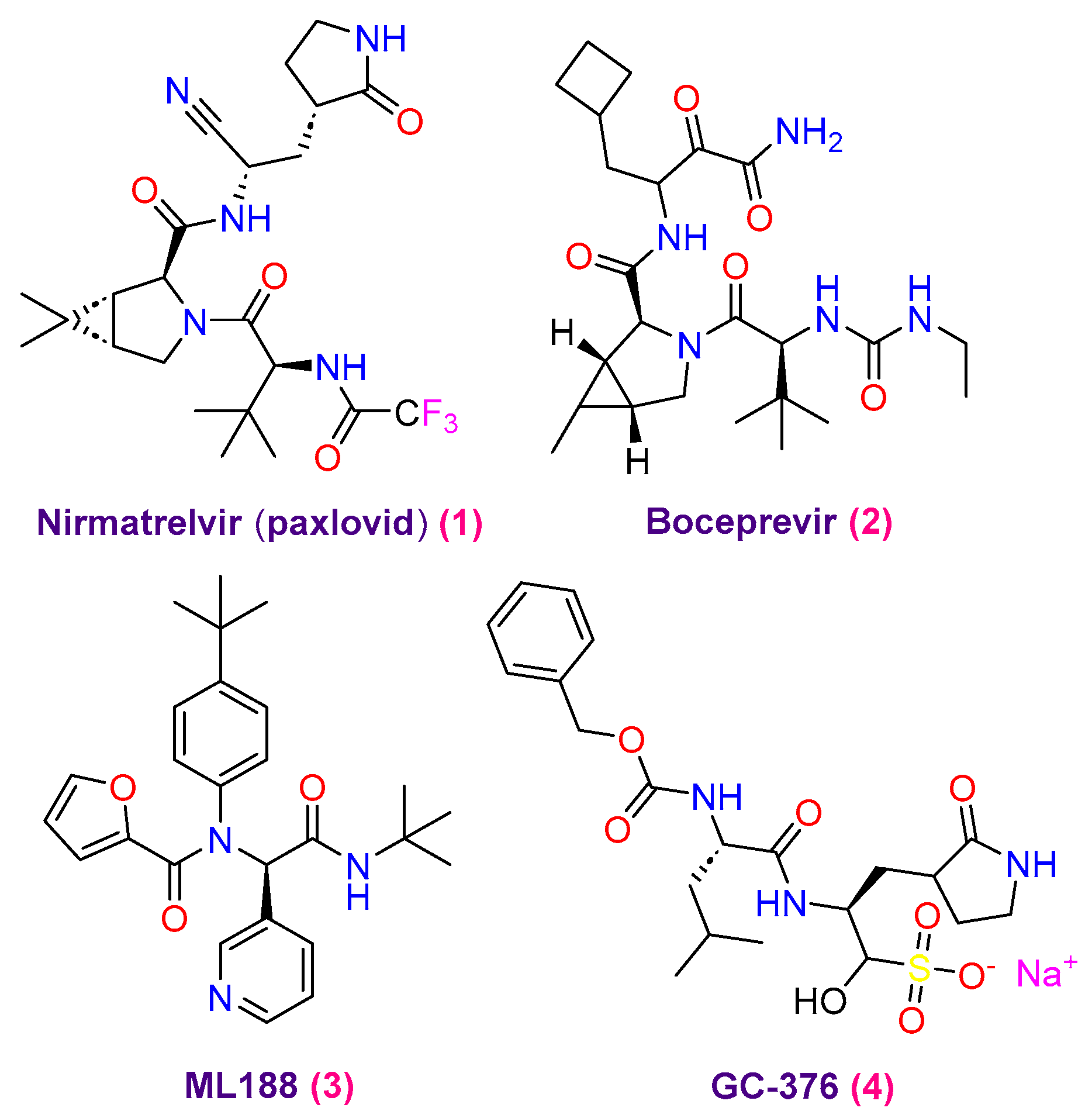
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.; Shahabi, D.; Chapman, M.E.; Mesecar, A.D. Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and COVID-19 Therapeutics. Chem. Med. Chem. 2020, 15, 907–932. [Google Scholar] [CrossRef]
- Kitamura, N.; Sacco, M.D.; Ma, C.; Hu, Y.; Townsend, J.A.; Meng, X.; Zhang, F.; Zhang, X.; Ba, M.; Szeto, T.; et al. Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2021, 10, 1021. [Google Scholar] [CrossRef]
- Zhang, M.; Wei, W.; Peng, C.; Ma, X.; He, X.; Zhang, H.; Zhou, M. Discovery of novel pyrazolopyrimidine derivatives as potent mTOR/HDAC bi-functional inhibitors via pharmacophore-merging strategy. Bioorg. Med. Chem. Lett. 2021, 49, 128286. [Google Scholar] [CrossRef] [PubMed]
- Tuyen, N.T.; Maged, H. Synthesis and Applications of Nitrogen-Containing Heterocycles as Antiviral Agents. Molecules 2022, 27, 2700. [Google Scholar] [CrossRef]
- Darabi, M.; Nikoorazm, M.; Tahmasbi, B.; Ghorbani-Choghamarani, A. Immobilization of Ni (ii) complex on the surface of mesoporous modified-KIT-6 as a new, reusable, and highly efficient nanocatalyst for the synthesis of tetrazole and pyranopyrazole derivatives. RSC Adv. 2023, 13, 12572–12588. [Google Scholar] [CrossRef]
- Almaghrabi, M.; Musa, A.; Aljohani, A.K.B.; Ahmed, H.E.A.; Alsulaimany, M.; Miski, S.F.; Mostafa, E.M.; Hussien, S.; Parambi, D.G.T.; Ghoneim, M.M.; et al. Introducing of novel class of pyrano[2,3-c] pyrazole-5-carbonitrile analogs with potent antimicrobial activity, DNA gyrase inhibition, and prominent pharmacokinetic and CNS toxicity profiles supported by molecular dynamic simulation. J. Biomol. Struct. Dyn 2023. Online ahead of print. [Google Scholar] [CrossRef]
- El-Sayed, M.K.F.; El-Shahawi, M.M.; Ali, Y.M.; Abdel-Haleem, D.R.; El-Azm, F.S.M.A. Synthesis, larvicidal efficiency and molecular docking studies of novel annulated pyrano [2,3-c] pyrazoles against Culex pipiens L. and Musca domestica L. larvae. Bioorg. Chem. 2023, 130, 106258. [Google Scholar] [CrossRef]
- Biswas, S.K.; Das, D. One-pot synthesis of pyrano [2,3-c] pyrazole derivatives via multicomponent reactions (MCRs) and their applications in medicinal chemistry. Mini Rev. Org. Chem. 2022, 19, 552–568. [Google Scholar] [CrossRef]
- Metwally, N.H.; Koraa, T.H.; Sanad, S.M.H. Green one-pot synthesis and in vitro antibacterial screening of pyrano [2,3-c] pyrazoles, 4 H-chromenes and pyrazolo [1,5-a] pyrimidines using biocatalyzed pepsin. Synth. Commun. 2022, 52, 1139–1154. [Google Scholar] [CrossRef]
- Ali, T.E.; Assiri, M.A.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I. Facile Green One-Pot Synthesis and Antiproliferative Activity of Some Novel Functionalized 4-(4-Oxo-4 H-chromen-3-yl) pyrano [2,3-c] pyrazoles and 5-(4-Oxo-4 H-chromen-3-yl) pyrano [2, 3-d] pyrimidines. Russ. J. Org. Chem. 2022, 58, 106–113. [Google Scholar] [CrossRef]
- Parikh, P.H.; Timaniya, J.B.; Patel, M.J.; Patel, K.P. Microwave-assisted synthesis of pyrano [2,3-c]-pyrazole derivatives and their anti-microbial, anti-malarial, anti-tubercular, and anti-cancer activities. J. Mol. Struct. 2022, 1249, 131605. [Google Scholar] [CrossRef]
- Mahmoudi, Z.; Ghasemzadeh, M.A.; Kabiri-Fard, H. Fabrication of UiO-66 nanocages confined brønsted ionic liquids as an efficient catalyst for the synthesis of dihydropyrazolo [4′,3′:5,6] pyrano [2,3-d] pyrimidines. J. Mol. Struct. 2019, 1194, 1–10. [Google Scholar] [CrossRef]
- Vahedi, M.M.; Asghari, S.; Tajbakhsh, M.; Mohseni, M.; Khalilpour, A. One-pot three-component synthesis of novel pyrano [3,2-e] pyrazolo [1,5-a] pyrimidines and investigation of their biological activities. J. Mol. Struct. 2023, 1284, 135446. [Google Scholar] [CrossRef]
- Ravindar, L.; Hasbullah, S.A.; Rakesh, K.P.; Hassan, N.I. Pyrazole and pyrazoline derivatives as antimalarial agents: A key review. Eur. J. Pharm. Sci. 2023, 183, 106365. [Google Scholar] [CrossRef]
- Becerra, D.; Abonia, R.; Castillo, J.-C. Recent applications of the multicomponent synthesis for bioactive pyrazole derivatives. Molecules 2022, 27, 4723. [Google Scholar] [CrossRef]
- Sadeghian, Z.; Bayat, M.; Safari, F. Synthesis and in vitro anticancer activity evaluation of spiro [indolo [2,1-b] quinazoline-pyrano [2,3-c] pyrazole] via sequential four-component reaction. J. Mol. Struct. 2022, 1250, 131759. [Google Scholar] [CrossRef]
- Talaiefar, S.; Habibi-Khorassani, S.M.; Shaharaki, M. Comprehensive kinetics and a mechanistic investigation on the biological active pyrano [2,3-c] pyrazole core in the presence of both eco-friendly catalyst and solvent: Experimental green protocol. Polycycl. Aromat. Compd. 2022, 42, 791–814. [Google Scholar] [CrossRef]
- Das, S.; Akbar, S.; Ahmed, B.; Dewangan, R.P.; Iqubal, M.K.; Iqubal, A.; Chawla, P.; Pottoo, F.H.; Joseph, A. Recent advancement of pyrazole scaffold based neuroprotective agents: A review. CNS Neurol. Disord. Drug Targets 2022, 21, 940–951. [Google Scholar] [CrossRef]
- Conti, C.; Monaco, L.P.; Desideri, N. 3-Phenylalkyl-2H-chromenes and -chromans as novel rhinovirus infection inhibitors. Bioorg. Med. Chem. 2017, 25, 2074–2083. [Google Scholar] [CrossRef]
- Addoum, B.; El Khalfi, B.; Sakoui, S.; Derdak, R.; Sakoui, S.; Elmakssoudi, A.; Soukri, A. Synthesis, in vitro Antimicrobial Activity, and Docking Studies of some Pyrano [2,3-c] Pyrazole Derivatives. Biointerface Res. Appl. Chem. 2022, 12, 4705–4730. [Google Scholar] [CrossRef]
- Mahmoud, A.; Mostafa, A.; Al-Karmalawy, A.A.; Zidan, A.; Abulkhair, H.S.; Mahmoud, S.H.; Shehata, M.; Elhefnawi, M.M.; Ali, M.A. Telaprevir is a potential drug for repurposing against SARS-CoV-2: Computational and in vitro studies. Heliyon 2021, 7, e07962. [Google Scholar] [CrossRef]
- Raj, V.; Lee, J.-H.; Shim, J.-J.; Lee, J. Antiviral activities of 4H-chromen-4-one scaffold-containing flavonoids against SARS–CoV–2 using computational and in vitro approaches. J. Mol. Liq. 2022, 353, 118775. [Google Scholar] [CrossRef]
- Amer, M.M.; Abdellattif, M.H.; Mouneir, S.M.; Zordok, W.A.; Shehab, W.S. Synthesis, DFT calculation, pharmacological evaluation, and catalytic application in the synthesis of diverse pyrano [2,3-c] pyrazole derivatives. Eur. J. Med. Chem. 2021, 114, 105136. [Google Scholar] [CrossRef]
- Mor, S.; Khatri, M.; Punia, R.; Nagoria, S.; Sindhu, S. A new insight into the synthesis and biological activities of pyrazole based derivatives. Mini Rev. Org. Chem. 2022, 19, 717–778. [Google Scholar] [CrossRef]
- Cheke, R.S.; Narkhede, R.R.; Ambhore, J.P.; Shinde, S.D. The Molecular Docking Study of Potential Drug Candidates Showing Anti-COVID-19 Activity by Exploring Therapeutic Targets of SARS-CoV-2. EJMO 2020, 4, 185–195. [Google Scholar] [CrossRef]
- El-Assaly, S.A.; Ismail, A.E.H.A.; Bary, H.; Abouelenein, M.G. Synthesis, molecular docking studies, and antimicrobial evaluation of pyrano [2,3-c]pyrazole derivatives. Curr. Chem. Lett. 2021, 10, 309–328. [Google Scholar] [CrossRef]
- Abouelenein, M.G.; Ismail, A.E.H.A.; Aboelnaga, A.; Tantawy, M.A.; El-Ebiary, N.M.; El-Assaly, S.A. Synthesis, DFT calculations, In silico studies, and biological evaluation of pyrano [2,3-c] pyrazole and pyrazolo [4′,3′:5,6] pyrano [2,3-d] pyrimidine derivatives. J. Mol. Struct. 2023, 1275, 134587. [Google Scholar] [CrossRef]
- Kumar, Y.; Harvijay, S.; Chirag, N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef]
- Debnath, P.; Bhaumik, S.; Sen, D.; Muttineni, R.K.; Debnath, S. Identification of SARS-CoV-2 Main Protease Inhibitors Using Structure Based Virtual Screening and Molecular Dynamics Simulation of DrugBank Database. ChemistrySelect 2021, 6, 4991–5013. [Google Scholar] [CrossRef]
- Ahmad, I.; Kuznetsov, A.E.; Pirzada, A.S.; Alsharif, K.F.; Daglia, M.; Khan, H. Computational pharmacology and computational chemistry of 4-hydroxyisoleucine: Physicochemical, pharmacokinetic, and DFT-based approaches. Front. Chem. 2023, 11, 1145974. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Yousef, R.G.; Elkady, H.; Gobaara, I.M.; Alsfouk, B.A.; Husein, D.Z.; Ibrahim, I.M.; Metwaly, A.M.; Eissa, I.H. Design, synthesis, docking, DFT, MD simulation studies of a new nicotinamide-based derivative: In vitro anticancer and VEGFR-2 inhibitory effects. Molecules 2022, 27, 4606. [Google Scholar] [CrossRef]
- AbouAitah, K.; Allayh, A.K.; Wojnarowicz, J.; Shaker, Y.M.; Swiderska-Sroda, A.; Lojkowski, W. Nanoformulation Composed of Ellagic Acid and Functionalized Zinc Oxide Nanoparticles Inactivates DNA and RNA Viruses. Pharmaceutics 2021, 13, 2174. [Google Scholar] [CrossRef]
- Ragab, S.S.; Sweed, A.M.K.; Elrashedy, A.A.; Allayeh, A.K. Design, Synthesis, Antiviral Evaluation, and Molecular Dynamics Simulation Studies of New Spirocyclic Thiopyrimidinones as Anti HCoV-229E. Chem. Biodivers. 2022, 19, 202200632. [Google Scholar] [CrossRef]
- Mostafa, A.; Kandeil, A.; Elshaier, Y.A.M.M.; Kutkat, O.; Moatasim, Y.; Rashad, A.A.; Shehata, M.; Gomaa, M.R.; Mahrous, N.; Mahmoud, S.H.; et al. FDA-Approved Drugs with Potent In Vitro Antiviral Activity against Severe Acute Respiratory Syndrome Coronavirus 2. Pharmaceuticals 2020, 13, 443. [Google Scholar] [CrossRef]
- ElNaggar, M.H.; Elgazar, A.A.; Gamal, G.; Hamed, S.M.; Elsayed, Z.M.; El-Ashrey, M.K.; Abood, A.; El Hassab, M.A.; Soliman, A.M.; El-Domany, R.A.; et al. Identification of sulphonamide-tethered N-((triazol-4-yl) methyl) isatin derivatives as inhibitors of SARS-CoV-2 main protease. J. Enzym. Inhib. Med. Chem. 2023, 38, 2234665. [Google Scholar] [CrossRef]
- Abo Elmaaty, A.; Eldehna, W.M.; Khattab, M.; Kutkat, O.; Alnajjar, R.; El-Taweel, A.N.; Al-Rashood, S.T.; Abourehab, M.A.; Binjubair, F.A.; Saleh, M.A.; et al. Anticoagulants as potential SARS-CoV-2 Mpro inhibitors for COVID-19 patients: In vitro, molecular docking, molecular dynamics, DFT, and SAR studies. Int. J. Mol. Sci. 2022, 23, 12235. [Google Scholar] [CrossRef]
- Said, M.A.; Albohy, A.; Abdelrahman, M.A.; Ibrahim, H.S. Remdesivir analog as SARS-CoV-2 polymerase inhibitor: Virtual screening of a database generated by scaffold replacement. RSC Adv. 2020, 12, 22448–22457. [Google Scholar] [CrossRef]
- Said, M.A.; Albohy, A.; Abdelrahman, M.A.; Ibrahim, H.S. Importance of glutamine 189 flexibility in SARS-CoV-2 main protease: Lesson learned from in silico virtual screening of ChEMBL database and molecular dynamics. Eur. J. Pharm. Sci. 2021, 160, 105744. [Google Scholar] [CrossRef]
- Chakraborti, S.; Bheemireddy, S.; Srinivasan, N. Repurposing drugs against the main protease of SARS-CoV-2: Mechanism-based insights supported by available laboratory and clinical data. Mol. Omics 2020, 16, 474–491. [Google Scholar] [CrossRef]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, 271–276. [Google Scholar] [CrossRef]
- Mesli, F.; Ghalem, M.; Daoud, I.; Ghalem, S. Potential inhibitors of angiotensin converting enzyme 2 receptor of COVID-19 by Corchorus olitorius Linn using docking, molecular dynamics, conceptual DFT investigation and pharmacophore mapping. J. Biomol. Struct. Dyn. 2022, 40, 7311–7323. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. Available online: http://www.jcheminf.com/content/4/1/17 (accessed on 28 June 2023). [CrossRef]
- Abouelenein, M.G.; El-Rashedy, A.A.; Awad, H.M.; El Farargy, A.F.; Nassar, I.F.; Nassrallah, A. Synthesis, molecular modeling Insights, and anticancer assessment of novel polyfunctionalized Pyridine congeners. Bioorg. Chem. 2023, 141, 106910. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Code | IC50 (µM) | SI (µM) |
|---|---|---|
| 1 | 61.878 | 3.5 |
| 2 | 309.059 | 2.4 |
| 3 | 80.308 | 2.1 |
| 4 | 70.115 | 0.9 |
| 5 | 51.239 | 1.86 |
| 6 | 89.385 | 7.6 |
| 7 | 25.761 | 4.3 |
| 8 | 69.106 | 0.6 |
| 9 | 133.933 | 2.3 |
| 10 | 19.474 | 1.3 |
| 11 | 10.908 | 1.7 |
| 12 | 18.593 | 3.5 |
| 13 | 88.784 | 0.46 |
| 14 | 78.729 | 6.5 |
| 15 | 123.451 | 1.18 |
| 16 | 58.056 | 1.05 |
| 17 | 95.390 | 1.7 |
| 18 | 44.862 | 12.6 |
| Compound | Mpro % Inhibition | S-Score (kcal/mol) | Involved Receptor Residues | Type of Bond Interaction |
|---|---|---|---|---|
| 6 | 80.4% | −6.1855 | Met165 Cys145 His163 Asn142 | H-bond H-bond H-bond H-bond |
| 7 | 73.1% | −5.8237 | Cys145 Met654 Gln189 | H-bond Hydrophobic Interaction Hydrophobic Interaction |
| 14 | 81.4% | −6.5673 | Met 165 Glu166 Cys145 Gln189 | H-bond H-bond H-bond Hydrophobic Interaction |
| 18 | 84.5% | −7.3091 | His 164 Gln189 Cys145 Asp187 Glu166 | H-bond H-bond H-bond H-bond Hydrophobic Interaction |
| Parameters | Compound 18 |
|---|---|
| EHOMO | −0.29094 |
| ELUMO | −0.17144 |
| Energy Gap (Egab) | 0.1195 |
| Dipole Moment (Dm) | 2.2848697 |
| Ionization Potential (IP) | 0.29094 |
| Electron Affinity (EA) | 0.17144 |
| Electronegativity (χ) | −0.1195 |
| Chemical Potential (µ) | 0.23119 |
| Global Chemical Hardness (դ) | 0.1195 |
| Global Chemical Softness (σ) | 8.2682 |
| Electrophilicity Index (Ѡ) | 0.223635 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allayeh, A.K.; El-boghdady, A.H.; Said, M.A.; Saleh, M.G.A.; Abdel-Aal, M.T.; Abouelenein, M.G. Discovery of Pyrano[2,3-c]pyrazole Derivatives as Novel Potential Human Coronavirus Inhibitors: Design, Synthesis, In Silico, In Vitro, and ADME Studies. Pharmaceuticals 2024, 17, 198. https://doi.org/10.3390/ph17020198
Allayeh AK, El-boghdady AH, Said MA, Saleh MGA, Abdel-Aal MT, Abouelenein MG. Discovery of Pyrano[2,3-c]pyrazole Derivatives as Novel Potential Human Coronavirus Inhibitors: Design, Synthesis, In Silico, In Vitro, and ADME Studies. Pharmaceuticals. 2024; 17(2):198. https://doi.org/10.3390/ph17020198
Chicago/Turabian StyleAllayeh, Abdou K., Aliaa H. El-boghdady, Mohamed A. Said, Mahmoud G. A. Saleh, Mohammed T. Abdel-Aal, and Mohamed G. Abouelenein. 2024. "Discovery of Pyrano[2,3-c]pyrazole Derivatives as Novel Potential Human Coronavirus Inhibitors: Design, Synthesis, In Silico, In Vitro, and ADME Studies" Pharmaceuticals 17, no. 2: 198. https://doi.org/10.3390/ph17020198