
Abstract
Although global vaccination campaigns alleviated the SARS-CoV-2 pandemic in terms of morbidity and mortality, the ability of the virus to originate mutants may reduce the efficacy of vaccines, posing a serious risk of a renewed pandemic. There is therefore a need to develop small molecules capable of targeting conserved viral targets, such as the main protease (Mpro). Here, a series of benzisoselenazolones and diselenides were tested for their ability to inhibit Mpro; then the most potent compounds were measured for antiviral activity in vitro, and the mechanism of action was investigated. Density functional theory calculations, molecular docking and molecular dynamics simulations were also used to elucidate the protein/drug interaction. Finally, a bio-organic model was established to study the reaction between selenorganic compounds and biologically relevant thiols to unveil possible metabolic pathways of such compounds. The overall results contribute to the identification of a series of novel Se-containing molecules active against SARS-CoV-2 and to the clarification of some important aspects in the mechanisms of action of such inhibitors targeting SARS-CoV-2 Mpro.
Similar content being viewed by others


Introduction
The Coronaviridae family was first globally recognized as a public health threat with the emergence of the severe acute respiratory syndrome (SARS) in 20031 and its potential was confirmed a decade later with the emergence of Middle East respiratory syndrome (MERS)2. However, it was the unprecedented rapid emergence of SARS-CoV-2 and the resulting COVID-19 pandemic that triggered global research effort to develop vaccines, therapeutics, and diagnostics to contrast this threat. Although effective vaccines are now available, they do not prevent the infection and additional layers of protection are still desirable in high-risk individuals. Unfortunately, despite the identification of several molecular targets of SARS-CoV-23,4,5, as well as human proteins involved in the viral entry and spread of the coronavirus that may represent further pharmacological targets6,7, the number of compounds required to drive an effective drug discovery program remains relatively limited compared to the undergoing efforts. In fact, most of the research has focused on drug repurposing8,9, with only nirmatrelvir having been developed as a novel antiviral medication on the basis of the earlier clinical candidate lufotrelvir10, and other efforts are currently still ongoing11,12.
Main protease (Mpro) and papain-like protease (PLpro) are two viral enzymes essential for virus replication and evasion of the host immune responses. The absence of closely related cellular homologs makes these enzymes particularly attractive targets for the design of coronavirus-specific antiviral agents. Among the first compounds to be discovered to block their enzymatic activity, the organoselenium compound 2-phenyl-1,2-benzoisoselenazol-3-one (ebselen, compound 1a)13,14 was identified as a potent inhibitor of Mpro, in a high-throughput screening study among a pool of existing small molecules15. Ebselen showed a potent inhibitory activity toward Mpro, with an IC50 in the high nanomolar range (670 nM) and inhibitory activity toward the infectious virus with an EC50 in the low micromolar range (4.67 µM)15. Subsequently, it was demonstrated that ebselen also inhibits the papain-like protease (PLpro) from SARS-CoV-1 and SARS-CoV-2 (with an IC50 = 2.26 µM for PLpro of SARS-CoV-2)16. The same authors of this seminal study recently reported a series of ebselen analogues as potent inhibitors of nsp14 guanine N7‑methyltransferase17.
Studies aimed at identifying the mode of action of ebselen (1a) suggested that it acts as an irreversible inhibitor by covalently binding to the reactive cysteines in the active sites of PLpro and Mpro to form a stable selenyl sulfide. Tandem mass spectrometry and computational calculations showed that for SARS-CoV-2 the reactive cysteine of Mpro is Cys1454,18, while molecular modelling suggested that Cys111 is the target of ebselen within the PLpro active site19. In addition, molecular dynamics simulations suggested that, in addition to covalent inhibition, ebselen may also interact and inhibit Mpro also non-covalently by binding to a pocket located between the II and the III domains of the protein18.
The ability of ebselen to readily modify cysteines has been well documented20,21,22,23,24, and this could be the the major problem for its actual clinical use. On the other hand, several studies have demonstrated an almost complete absence of toxic effects in vivo, and this has also been observed in the clinical trials in which 1a has been evaluated14,25. In addition, from a general point of view, ebselen has several additional pharmacological effects that have been shown to be favorable in the context of COVID-19 healing26.
Apart from ebselen, only a few other selenium-containing compounds with anti-SARS-CoV-2 activity have been reported in the literature (Fig. 1).

Some Se-containing compounds endowed with anti-Mpro and anti-SARS-CoV-2 activity. IC50 denotes the concentration at which enzyme activity is reduced by 50%; EC50 denotes the concentration at which the viral replication in Vero cells is reduced by 50%, as determined by RT-qPCR.
In particular, Yang, Zhang, O’Neill and Hasnain tested a series of benzisoselenazolone analogs, that had previously been reported as neuroprotective agents. They identified compounds 1b and 1c as more active than 1a in inhibiting Mpro. At the same time, aiming at resolving the co-crystal structure with the protein, the authors reported that these compounds were able to transfer a hydrogenselenide unit to Cys145 through an SNAr-like reaction that takes place in the Mpro active site27. A related study was carried out by Kumar et al., which demonstrated the selenylation of the same cysteine using other benzisoselenazolones28. Recently, Rana et al.. reported a series of ebselen close analogues with anti-Mpro activity, with compounds 1d and 1e being the best in class. These derivatives showed an improved antiviral activity not only in Vero cells but also in other cell lines chosen because they better mirrored the lung epithelium29. Zhang, Wang, et al. recently reported benzisoselenazolones bearing different aromatics at the N2 positions. In their study, authors identified compounds endowed with low nanomolar IC50 and antiviral activity in the micromolar range30.
Besides benzisoselenazolones, selenides proved to be suitable substituents to enhance the anti-SARS-CoV-2 activity of the natural product quercetin31. The resulting selenoquercetin analogs (2a and 2b) showed a striking ability to inhibit Mpro, with compound 2b also being active in vitro with an EC50 in the low micromolar range and without showing any cytotoxicity32. More recently also other selenides have been reported by some of us to be able to inhibit SARS-CoV-2 in a cellular context33 Antiviral activity (in vitro CC50 (24.61 µM) and EC50 (2.39 µM)) was reported also for (PhSe)2 which was recognized as a good inhibitor of the SARS-CoV-2 virus replication in a cell culture model; the inhibition mechanisms were also described for this archetypal diselenide in silico through a combined molecular dynamics (MD) and density functional theory (DFT) approach34.
Taken together, these results clearly highlight the value of selenorganic derivatives, in particular ebselen-like structures, as lead compounds for the development of novel anti-SARS-CoV-2 agents.
With this in mind, we herein report the anti-SARS-CoV-2 properties of ebselen-like derivatives (compounds 1f-s) and a series of closely related diselenides (compounds 3,4). Considering that in studies reporting an anti-SARS-CoV-2 activity the ebselen core is always decorated with aromatic and benzylic substituents, we sought to prepare benzisoselenazolones bearing aliphatic side chains to extend the structure-activity relationship (SAR) for this class of compounds. In addition, we provide mechanistic insights to prove that, for certain derivatives, a plausible metabolic link between benzisoselenazolones and the corresponding diselenides does indeed exist.
For the whole set of compounds herein reported, the anti-Mpro activity was initially assessed through an in vitro screening, and then mass spectrometry confirmed the covalent enzymatic inhibition. The most potent derivatives were then tested in a cellular model of SARS-CoV-2 infection, by investigating also their mechanism of action. Not only benzisoselenazolones but also diselenides were found to be active. A bioorganic, NMR-based model was purposely developed to get insight into the intracellular fate of the studied compounds.
Results and discussion
Synthesis and characterization
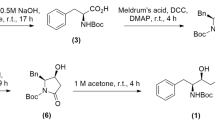
Diphenyl diselenide (8) and selenocystine (6) are commercially available. Dibenzyl diselenide (7) was prepared following the procedure reported in literature35. Ebselen-like compounds 1f-1 L were prepared starting from 535,36, which was first coupled with amino acids protected as esters leading to compounds 3f, h-l.37 The ester derivatives were then converted into the corresponding benzisoselenazolone 1f-l, following a procedure recently reported by some of us38. The diselenides with an acid moiety were obtained starting from the corresponding esters by mild basic hydrolysis, leading to compounds 4f, h-l (Fig. 2).

Diselenides 3n-q, s were prepared from the corresponding ebselen-like compounds (1n-q, s)39 by sequential NaBH4-mediated reduction of the Se-N bond and air oxidation of the so-formed selenolate anion (Fig. 3)40.

Synthesis of diselenides 3n-q, s.
As a first line of screening, the whole set of compounds was preliminarily assayed in vitro at the concentration of 40 µM, which showed complete inhibition of the enzymatic activity of Mpro (see Table S1). The IC50 was then determined, and the results are collected in Tables 1, 2 and 3.
The ebselen derivatives (1f-s) were generally more potent than the corresponding diselenides 3f-s and 4f-l. Derivatives 1k and 1i were the best in class among the compounds tested, with an IC50 in the low nanomolar range. In all the cases, the anti-Mpro activity was higher than that of ebselen and most of its derivatives reported in the literature, highlighting that the replacement of the aromatic ring with an aliphatic one improves the ability to bind and inhibit the viral protease. Conversely, the terpene containing 1 m-s derivatives showed activity in the high nanomolar range.
Regarding the diselenides, compounds 5 and 7 were unable to exert any sort of inhibition, whereas selenocystine (compound 6) had an IC50 in the high micromolar range. Also, diphenyl diselenide 8 showed a low micromolar activity. The introduction of a carboxamide substituent in the ortho position with respect to the selenium atom preserved or even improved the activity, as in the case of compound 3f. From the comparison among compounds containing an ester functionality (3 h-l) with the ones containing an acid moiety (4 h-l), it clearly emerged that the latter were less active in the Mpro inhibition. Again, the terpene derivatives 3p-q and 3s were able to inhibit Mpro, but they required a higher concentration compared to the other compounds, and compound 3n was completely inactive. Based on these data, a structure activity relationship (SAR) summary was attempted and is illustrated in Fig. 4.

SAR summary based on the data of Mpro inhibition of both benzisoselenazolones and diselenides.
The better activity of ebselen-derivatives in comparison to the diselenides could be ascribed to a higher electrophilicity of the former, which facilitates the reaction with reactive thiols and, consequently, the covalent inhibition of the protein. The greater electron deficiency of ebselen derivatives can be clearly deduced from the chemical nature of the two functional groups (a selenazolone and a diselenide) and is also confirmed spectroscopically by comparing their76Se NMR chemical shifts, which range from 804 to 935 ppm for benzisoselenazolones and from 439 to 450 ppm for the diselenides.
Molecular modeling
Computational techniques including density functional theory (DFT) quantum simulations, classical (rigid/flexible) molecular docking, and molecular dynamics simulations were also used to model the binding of our compounds to the main protease Mpro. As a further evaluation of the electrophilicity of these compounds, the geometry of some selected ebselen-like and diselenides systems was optimized by DFT (B3LYP-D3/def2-TZVP level of theory), and the natural partial atomic charges were computed by using the Natural Population Analysis (NPA) as implemented in the NBO software suite. The results are shown in Table 4.
The q(Se) value resulted to be inversely proportional to the IC50 value; compounds having q(Se) > 0.610 e are the most active. This observation further supports the hypothesis of a nucleophilic attack of the sulfur of the cysteine to the selenium as a key step in the inhibition process in the tests in vitro. Noteworthy, some diselenides show an asymmetric charge distribution among the two selenium atoms. This is aconsequence of a non-symmetrical involvement of the selenium atoms in weak interactions with the nearby chemical environment (Figure S1). For example, as shown in Figure S2, in 3p only one selenium is involved in two hydrogen bonds with the two amide groups, producing a slight polarization of the Se-Se bond.
Molecular docking was also used as an additional computational tool to investigate the binding of selected ligands in the active site of Mpro, and the steric constraints that may limit their access to the binding pocket. In particular, we focused on 1a (ebselen), 1i, and 1r, as representative examples of compounds with subtle structural difference leading to appreciable variations among their IC50 values (see Table 1 and data reported in literature for 1a10). The results reported in Fig. 5; Table 5 show that these compounds anchors in the binding site of Mpro with a good affinity. The docking scores were at least ‒6.0 kcal/mol, which is comparable to the values previously found for other experimentally-validated binders of this protein (e.g., the same value was found for eugenol, and up to ‒7.2 kcal/mol for quercetin in a large screening campaign)31,41. The differences among the three compounds tested were negligible within the limit of the accuracy (about ± 0.3 kcal/mol) in the scoring function of the docking algorithm42. This finding suggests that the activity in the high nanomolar range observed for 1r, and in common with the other terpenes-containing derivatives, should not be ascribed to steric clashes within the binding site. We also observed that the affinities of these compounds did not change significantly by considering as flexible the side chains of protein residues His41/Cys145 (Table 5), indicating that rearrangements of the catalytic dyad of Mpro do not play an essential role in the binding.We also performed additional simulations by using as docking host the protein crystallographic structure obtained in the presence of an Ebselen-derivative inhibitor (PDB entry 7W9G), showing that the most favorable docking poses of our compounds are very similar (Figure S3).

Docking poses obtained for compounds 1a (ebselen), 1i, and 1r. A Superposition of all the docking poses, with the catalytic dyad His41/Cys145 labeled; B compounds 1a (cyan); C compound 1i (magenta); D compound 1r (green).
In the docking poses, the minimum non-bonding distance found between the sulfur atom of Cys145 and the selenium was ~ 3.7 Å, as it could be expected considering their van der Waals radii (1.8 and 1.9 Å, respectively). However, we also verified that each compound is able to form a covalent bond Se‒S (with a distance < 2.5 Å) and still fit within the active site of Mpro without requiring a local reorganization of the protein pocket. At variance with typical covalent docking studies on SARS-CoV-2 that have a more stringent predictive value on the binding location43, this was obtained by performing simulations were the bond was assumed as already formed, and verifying that the resulting adduct (encompassing the side chain of the catalytic residue Cys145 plus either of the compounds) could favourably self-dock in the protein active site.
Molecular dynamics simulations were used to corroborate the docking results. Trajectories were run for the three ligands starting from two docking poses in two crystal structures of Mpro (6E2Y and 7W9G PDB entries) with a total of 12 runs per ligand. Calculations of the binding energy show that for all systems there is a favorable affinity (see supplementary information Table S4 for the full list of binding energies). However, the presence of a negatively charged Cys residue showed also that in some of the replicas solvent molecules can enter the binding site and favorably interact with Cys145 resulting in the displacement of the ligand. Overall, the combined results of docking and molecular dynamics show that the protein-ligand non-covalent interaction might be intact until the formation of the covalent bond Se‒S. On the other hand, mechanistic details of the formation of the covalent bond in Mpro have been previously reported in two independent DFT studies44,45 which showed agreeing results despite being conducted on enzymatic clusters of different size.
Biological assays
We then evaluated whether the anti-Mpro activity of the most potent compounds translated into the inhibition of the SARS-CoV-2 replication. The selected compounds were glycine derivatives 1f, 1 g, 3f, and 4f, isoleucine derivatives 1i and 3i, valine derivatives 1k, 3k, and 4k, and benzisoselenazolones containing glutamic and aspartic esters 1j and 1k. Ebselen 1a was tested in parallel as a positive control. First, the cytotoxicity was evaluated at 100, 80, 60, 40, 20 µM concentration in Vero cells. No toxicity was observed for all compounds except for 3f that that was highly toxic at 100 µM (Figure S4). Next, confluent monolayers of Vero cells were infected with the virus in the presence or absence of the inhibitors. All compounds were tested at 100, 80, 60, 40, and 20 µM concentration (except for 3f, which was not tested at 100 µM). 2 days post-infection, the cytopathic effect was assessed, and toxicity for 1f, 1 L, 3i was observed at 100 µM and for 1 g at 100 and 80 µM, therefore these conditions were not collected for further analysis. Cytotoxicity for some compounds observed in infected cells as compared to XTT assay could be due to cellular stress caused by viral infection. All remaining samples were harvested and evaluated by means of quantitative PCR coupled with reverse transcription (RT-qPCR) (Figure S5). The EC50 values were determined as summarized in Table 6, with the exception of compound 4f, which was found to be inactive. All compounds inhibited viral replication at low micromolar levels, with the ebselen-like compounds 1j and 1 L endowed with the lowest, and mutually similar, EC50 (7 and 8 µM, respectively). From a SAR standpoint, the double ester functionality seems to improve the antiviral activity of the compounds. The similarity of the EC50 of the two compounds mirrors their activity toward the Mpro, against which they showed comparable potencies (see Tables 1 and 6). Compound 1i, which is a nanomolar Mpro inhibitor, inhibits the viral replication with remarkable activity. Unexpectedly, the most potent Mpro inhibitor, 1k, exerted an antiviral activity lower than expected based on the biochemical assay, most probably for pharmacokinetic reasons. Among diselenides, the esters were way more potent than acids (see data for 3fvs.4f, and 3kvs.4k), regardless of their side chain.
Time of addition (TOA) experiments were also carried out to map the mode of action of the compounds. The most promising ebselen-like derivatives 1i and 1 L, and diselenides 3i and 3k, together with 1a, were added at three different stages of virus infection at a concentration of 60 µM.
Assay I (PRE) was used to assess whether the compounds may render the cells resistant to the virus. As such, they were added before infection and cells were pre-incubated with compounds for 1 h at 37 °C. Assay II (WITH) was to verify whether the compounds affect viral entry (i.e., early stages of replication). Thus, they were added with the virus, during the infection. Finally, the assay III (POST) in which compounds were added 2 h after infection was performed to assess their impact on viral replication and egress (i.e., late stages of replication).

TOA experiments for selected compounds. The inhibition of virus replication in Vero cells by tested compounds added at different times of infection (PRE, WITH and POST – see main text for description). The figure shows qRT-PCR analysis of cell culture supernatants infected with SARS-CoV-2 at TCID50 of 1600 per mL) 12 h post-infection. All the experiments were performed in triplicate, and the results are presented as averages, with error bars denoting Standard Error of the Mean (SEM). To determine the significance of the obtained results, Kruskal-Wallis multiple comparisons test was performed. P values of < 0.33 (*); <0.002 (**); <0.001 (***) were considered significant.
As shown in Fig. 6, the compounds 1i, 1j, and 1 L inhibited the SARS-CoV-2 virus at the early and late stages of the infection. We did not observe any effect of preincubation of cells or virions, suggesting that the inhibition occurs during the virus entry and replication. In the case of compounds 3i, 3k, and for ebselen (1a) used as a control, inhibition was observed at multiple stages, including in the pre-incubation assay, suggesting a different mechanism of action or an intracellular accumulation of the compounds. Conversely, the lack of activity of ebselen-like compounds 1i, 1j, and 1 L could derive from their reaction/interaction with cellular thiols different from Cys145 of Mpro. Such a reaction would convert the compounds into their selenylsulfide analogs, whose activity against the viral protease remains to be proven.
The electrophilic reaction of selenium-containing compounds with nucleophilic thiols is a well-known process46, and this pro-oxidant property should explain why some of them are toxic. Among the thiols, glutathione (GSH) is the most abundant in living cells, especially under oxidative stress conditions47. For this reason, a direct interaction between every electrophilic selenium-containing compound and GSH may be considered as the first chemical event involving them in the cellular environment. With the aim to improve our understanding of this topic, we set up an NMR-based model meant to study the reaction/interaction between selected compounds and GSH. In particular, beside an1H-NMR analysis, we selected the76Se NMR which represents a simple and reliable method for the identification of organoselenium derivatives in unpurified reaction mixtures carried out directly in an NMR tube48. Unfortunately, limitations in terms of sensitivity and relaxation time hampered quantitative interpretation of the results.
The poor solubility of ebselen and its derivatives in buffered aqueous conditions forced us to choose DMSO-d6 as solvent for the reaction, although it cannot be considered completely inert due to its mild oxidising properties.
As reported by Back and coworkers, one molar equivalent of reduced glutathione rapidly converts 1 into 9 (Fig. 7)49. It is also known that the second equivalent of reductant (GSH) promotes the rapid and quantitative formation of the corresponding diselenide 3a. We demonstrated that this latter transformation can be activated by 5 mol % of starting from 9 and leading to the formation of diselenide 3a and oxidized glutathione in up to 50% conversion, accounting for a self-catalytic process according to the equilibrium depicted in Scheme 3 (Figure S6).

Fate of ebselen 1a and the corresponding diselenide 3a in the presence of GSH.
Diselenide 3a, synthesized according to literature16, was analyzed by76Se NMR in DMSO-d6 and a peak at 442 ppm was observed. By the addition of a one molar equivalent of GSH, the signal of selenylsulfide 9 appeared at 547 ppm. Not all of the diselenide was consumed in the reaction, indicating that the reaction with GSH is not as fast as that with compound 1 (Figure S7). The diselenide 3k was subjected to the same investigations and showed a similar behaviour to the derivative 3a (Figure S8).
Two things are clear from the proposed mechanism: (1) under reducing conditions ebselen 1a cannot be considered as the final chemical entity responsible for the interaction with Mpro; (2) both 1a and the corresponding diselenide 3a in the presence of a reducing thiol establish an equilibrium between 9, 10 and 3a, that it was demonstrated to be prone to further thiol exchange processes. When this mixture was reacted with N-acetylcysteine (NAC), 76Se NMR showed the formation of another compound having a chemical shift compatible with that of the selenylsulfide 11 (Figure S9). This indicate that the above-described equilibria can be further modulated by thiol exchange processes.
These data suggest that special care should be taken when interpreting the data obtained from the enzymatic inhibition assay. Indeed, if it is carried out under non-reducing conditions, it may not be able to correctly interpret the molecular mechanism and take into account the molecular species actually involved in a real biological environment, thus losing its predictability.
For this reasons, freshly prepared compounds 1a and 3a were assayed in parallel against Mpro both in the presence and absence of DTT (as a reducing thiol). The results are summarized in Table 7.
As also reported by Wang50, the anti Mpro activity of 1a drops dramatically in the presence of DTT, and this is also true for diselenide 3a, that, in the presence of the reducing agent, displays an IC50 in the high micromolar range. As expected, in the absence of a reducing agent, ebselen is more potent that its diselenide, mirroring the higher electrophilicity of the selenium atom. In the presence of DTT, both 1a and 3a produced the same effect in terms of IC50 (considering that 3a is a dimer), in accordance with the mechanism proposed in Scheme 3.
Clearly, this mechanism require that that Se-Se bond should be polarized. As an example, glutathione is not able to reduce diphenyl diselenide 8 even when used in a large excess. In our cases, the amide functionality adjacent to the selenium atom can establish a non-bonded interaction able to modulate the electrophilicity and the redox properties of the selenium atom. This underlines that probably not all the diselenides (and not all the ebselen-derivatives) share the same reactivity and mechanism in the enzyme inhibition. Beside the covalent inhibition, a non-covalent mechanism of action can reasonably be speculated for some of them. This is the case of compound 8, which is still able to inhibit Mpro, but thought a non-covalent mechanism as recently proposed by Orian and Rocha34.
HRMS analysis performed under reducing conditions (for the presence of DTT, see SI) evidenced that both selenazolones and diselenides bind covalently the Mpro, but not in the same manner and not selectively. The mass of apo Mpro is ~ 33.8 kDa, in line with previous observations10. In experiments with ebselen 1a, and the analogous 1i, 1 L, and 1k, a mass corresponding to Mpro in covalent complexes with one, two, or three intact molecules was observed. In contrast, 3a and 3i, afforded peaks corresponding to Mpro in covalent complexes with one, two, or three halves of the intact compounds. In the case of 1j, the increment in Mpro mass did not correspond to either the intact molecule or half of it. Finally, 3k did not produced relevant covalent modification of Mpro.
To rule out that the inhibition could arise from compound 9, we tested by molecular docking simulations whether it could sterically bind within the active site of Mpro, but the results suggest that the GSH moiety would remain solvent-exposed, and sterically hamper the anchoring of the compound with the selenium in a position favorable to form a covalent bond with Cys145. This finding is coherent to the observations recently reported by Teixeira da Rocha51.
Finally, as a further investigation of the mechanism showed by those compounds that were proved to inhibit viral replication when administered before virus addition, an entry inhibition assay was set up. Compounds 1a, 3i, and 3k were tested for their ability to inhibit the spike (S) protein-mediated internalization of pseudoviruses, using the VSV-G-glycoprotein-decorated pseudoviruses as control. Such a system allows one for the identification of a direct interference with the entry process and the assessment of the selectivity of this process. As shown in Fig. 8, compound 1a showed the ability to block the entry of both pseudoviruses when tested at 60 µM, indicating that it interferes with the viral entry process in an unspecific manner. In contrast, compounds 3i and 3k did not inhibit the entry of the pseudoviruses.

Inhibition of SARS-CoV-2 pseudovirus entry. The inhibition of SARS-COV-2-S (A) or control VSV-G (B) pseudovirus (positive control) entry to HeLaACE2+ cells by 1a, 3i, and 3k compounds at 60 µM concentration. The figure shows normalized signal from cell culture lysates collected 72 h post-inoculation. All the experiments were performed in triplicate, and the results are presented with standard deviations (SD) error bars. The data were normalized to untreated transduced control which was arbitrary set at 100%; Kruskal-Wallis multiple comparisons test was performed to assess the significance of obtained results. P values of < 0.33 (*); <0.002 (**); <0.001 (***) were considered significant.
Conclusion
A series of new Mpro inhibitors were identified from the screening of some benzisoselenazolones and diselenide analogues. Based on computational studies, we may assess that the inhibition is likely to be covalent, with a direct proportionality between the IC50 and the electrophilicity of selenium among the tested compounds. Most of the Mpro inhibitors are endowed with antiviral activity measured in a cellular context without major cytotoxicity, thus leading to positive selectivity index values, even if the non-specific activity of selenium compounds is well-described and reported in several cases50,52,53,54. Time of addition studies proved that, although the antiviral potency (expressed in terms of EC50 values) is similar among the compounds, differences are still present. Ebselen, as an example, inhibits viral replication when added either pre-, with- and post–infection, whereas benzisoselenazolones 1i, 1j and 1 L are devoid of any activity when added pre-infection. This indicates that minor structural modification affects the antiviral properties but not the outcome of the enzymatic inhibition test.
We have also demonstrated that the antiviral activity cannot be traced back in a simplistic manner to the direct interaction of benzisoselenazolones with residue Cys145 of Mpro. Metabolism for sure plays a role as we proposed trough an NMR based bioorganic model in which the compounds are challenged with the most abundant cellular thiol, GSH. In the case of Ebselen, its marked electrophilicity leads it to a rapid and quantitative glutathionation, most likely affording a mixture in which the glutathionated adduct is in a dynamic equilibrium with the corresponding selenol and diselenide. This equilibrium can be obtained also starting from the diselenide, and it is prone to further modulation thought thiol exchange processes. This process could be responsible for the non-selective selenenylation of different free cysteines of the enzyme. Furthermore, it was demonstrated that ebselen also acts by inhibiting the spike (S) protein-mediated internalization of pseudoviruses, accounting for a multi-faced non-selective mechanism of action that involves both benzisoselenazolone derivatives and the corresponding diselenides. A more holistic approach is therefore necessary in the interpretation of the mechanism of action of these derivatives, and any reductionist simplification may miss important details of their biological activity.
Experimental section
DFT studies
All the geometry optimizations were performed with the Orca code55, version 4.1.0 at the B3LYP/def2-TZVP level and def2/J auxiliary basis. Dispersion effects were taken into account using the Grimme D3-parametrized empirical dispersion correction, with the Becke–Johnson (BJ) damping function56,57. Frequency calculations were carried out at the same level of theory, to ensure that the stationary structures had no imaginary frequencies. The solvent effects were modelled using the Conductor-Like Polarizable Continuum Model (CPCM), with water as solvent.
NBO atomic charges has been computed using the NBO6 suite of software58.
Molecular docking
Molecular docking was performed by using AutoDock Vina, version 1.2.3 (https://github.com/ccsb-scripps/AutoDock-Vina)42, following a protocol already used to test the binding of other ligands to the main protease of SARS-CoV-2 41,59. The structure of Mpro was taken from the crystal conformation of the ligand-free enzyme (Protein Data Bank entry: 6Y2E60) and considered either fully rigid or partly flexible in the sole side chains of the catalytic dyad His41/Cys145. Residue Cys145 was modelled as deprotonated, and His41 protonated solely at position Nδ. Compounds 1a, 1i, and 1r, simulated as representative examples of ebselen-like ligands, were built by using the molecular editor Avogadro, version 1.2.061, and full flexibility was allowed for rotations around their dihedral angles. The binding of Se-GSH to the active site was also probed in the same way. In all cases, a blind search was carried out within a volume encompassing the whole protein, and with an exhaustiveness 16 times larger than the default value62.
In addition to the simulations described above, further simulations were run to test whether compounds 1a, 1i, and 1r could be covalently bond with the catalytic Cys145 without requiring a structural reorganization of the active site of Mpro, as described below. In this case, Mpro was built from the complex with an ebselen-derivative inhibitor (Protein Data Bank entry: 7W9G62), in which the sole coordinates of the selenium atoms bound to S-Cys145 are reported. Since AutoDock Vina is not able to simulate a covalent docking, each of our compounds was modeled as an adduct already bound to the (modified) residue Cys145, and with the same Cα‒S‒Se geometry present in the crystallographic structure. Afterwards, the self-docking of the fully-flexible side chain of such Cys-modified residue and the rest of the rigid protein structure was simulated. The existence of bound conformations of the compounds that fit into the protein active site was verified by visual inspection, as well as by binding affinities (< ‒15 kcal/mol) incompatible with extensive steric clashes (which would lead instead to large positive values).
Molecular dynamics simulations
Partial atomic charges to model ligands 1a, 1i, and 1r were obtained with the antechamber software63 through a RESP fit of the electrostatic potential calculated on the HF/6-31G* optimized structures with Gaussian 1664. Bonded parameters were taken from the literature65 for the bonds and angles involving Se. All other protein and ligand parameters were taken from the FF14SB protein force field66. Molecular dynamics (MD) simulations of the compounds 1a, 1i, and 1r bound to Mpro were conducted usingGROMACS version 2023 − 2.68,69,70,71,72,73,74 Starting from the docked structures of the three ligands in two poses using two different crystal structures of Mpro (PDB entries 6Y2E and 7W9G), energy minimization with a 200 kJ mol− 1 nm− 1 maximum force tolerance was followed by 2 ns of NVT dynamics with a stochastic velocity rescale thermostat74. In the first ns of the simulation an annealing procedure was implemented to gradually take the temperature from 10 K to 298 K. Production runs were carried out for 200 ns using a stochastic velocity and cell rescaling algorithm in the NPT ensemble. In all MD runs the timestep was of 2 fs and a LINCS constraint was applied to all bonds involving hydrogen75. Protein-ligand binding energy calculations were conducted on the last 50 ns of the production runs using the MMPBSA.py.MPI script routine76 using a salt concentration of 0.1 M for the generalized Born model.
Chemistry
Compound 6 and 8 were purchased from Sigma Aldrich and used without further purifications. Compounds 1a, 1f-1 L were prepared as we previously reported in Nascimento et al.38, compound 3a was prepared as reported in Weglarz-Tomczak et al.16, compounds 3f, 3 h-3 L, 4f, 4 h-4 L and 5 were prepared as reported in Sancineto et al.37. Compound 7 was prepared as reported in Krasowska et al.35. Diselenides 3n-q, s were prepared from the corresponding ebselen-like compounds (1n-q, s)39 via a sequential NaBH4-mediated reduction of the Se-N bond and air oxidation of the so-formed selenolate anion40. Spectral data are superimposable to those reported in literature.
NMR experiments
NMR experiments were carried out at 25 °C on a Bruker Avance NEO 600 MHz spectrometer equipped with Cryoprobe Prodigy and operating at 600 MHz for 1H, and 114.45 MHz for 76Se experiments. 1H, and 76Se chemical shifts (δ) are reported in parts per million (ppm) and they are relative to TMS 0.0 ppm and the residual solvent peak of DMSO-d6 at δ 2.35 ppm76. Se experiments were referenced to PhSe276Se δ = 463 ppm in CDCl3).
For the experiments reported in Figure S6, a solution of compound 9 (21 mg, 0.036 mmol), prepared as reported in Sands et al.49, in 1 ml of DMSO-d6 was prepared, then 0.0018 mmol of GSH (from a 0.4 M stock solution in D2O) were added. Sequential 1H and 76Se NMR spectra were recorded.
For the experiment reported in Figure S7, a solution of compound 3a (10 mg, 0.018 mmol) in 1 ml of DMSO-d6 was prepared, then a stoichiometric amount of GSH (0.018 mmol from the above-mentioned stock solution) was added. Sequential 1H and 76Se NMR spectra were recorded.
For the experiment reported in Figure S8, a solution of 3k (12 mg, 0.018 mmol) in 1 ml of DMSO-d6 was prepared, then a stoichiometric amount of GSH (0.018 mmol from the above-mentioned stock solution) was added. Sequential 1H and 76Se NMR spectra were recorded.
For the experiment of Figure S9, a solution of 1a (5.1 mg, 0.018 mmol) in 1 ml of DMSO-d6 was prepared, then a stoichiometric amount of GSH (0.018 mmol from the above-mentioned stock solution) was added. Sequential 1H and76Se NMR spectra were recorded. To the same sample, a stoichiometric amount of N-acetyl cysteine (NAC, from a 0.4 M stock solution in D2O) was then added. Sequential 1H and 76Se NMR spectra were recorded to reveal the formation of a 1:1 mixture of two selenylsulfides 9 and 11. To the same sample, 0.018 mmol of GSH was later added, sequential1H and 76Se NMR spectra were recorded, and a higher amount of 9 was observed as revealed by the heights of the 76Se resonance peak. Finally, to the same sample, 0.036 mmol of NAC was added, and a higher amount of 11 was observed as revealed by the heights of the 76Se resonance peak.
Cells and viruses
Vero cells (Cercopithecus aethiops; kidney epithelial; ATCC CCL-81) and HeLa cells overexpressing ACE2 (HeLa[ACE2) cells were maintained in Dulbecco-modified Eagle’s medium (DMEM, high glucose, ThermoFisher Scientific, Poland) supplemented with 5% heat-inactivated fetal bovine serum (FBS, ThermoFisher Scientific, Poland). The medium was supplemented with penicillin (100 U/mL, ThermoFisher Scientific, Poland) and streptomycin (100 µg/ml, ThermoFisher Scientific, Poland). Cells were cultured at 37 °C in an atmosphere containing 5% CO2 and humidity. Every two weeks, cells were tested for mycoplasma contamination.
Reference SARS-CoV-2 strain 026 V-03883 was kindly granted by Christian Drosten, Charité—Universitätsmedizin, Berlin, Germany, by the European Virus Archive—Global (EVAg); https://www.european-virus-archive.com/, accessed on 15 April 2021).
SARS-CoV-2 stock was generated by infecting monolayers of Vero cells. The cells were incubated at 37 °C under 5% CO2. The virus-containing medium was collected at day 2 post-infection (p.i.), aliquoted, and stored at -80℃. Control samples from mock-infected cells were prepared in the same manner.
Virus yields were assessed by titration on fully confluent cells in 96-well plates according to the method of Reed and Muench77. Plates were incubated at 37℃, and the cytopathic effect (CPE) was scored by observation under an inverted microscope.
Time of addition assay
To discover the mechanism of action of the tested compounds, the inhibitors were added at three different stages of virus infection: PRE – compounds were added before infection, and cells were pre-incubated with compounds for 1 h at 37 °C; WITH – compounds were added to the virus, during the infection; POST – compounds were added after infection (2 h post-infection). In details, the Vero cells were seeded in a culture medium on a 96-well plate 1 day before infection. Fully confluent cells were inoculated with 1600 TCID50/ml of the SARS-CoV-2 virus, either in the presence or absence of the inhibitors. Mock control and medium control were also included. Cells were then incubated for 2 h at 37 °C and 5% CO2. Afterward, the cells were washed twice with PBS. Only in the POST-treatment version, each compound was applied into the cells monolayer, whereas in other conditions (PRE and WITH) the medium was applied into the cells monolayer. Cell culture supernatants were collected after 24 h for viral RNA isolation. The SARS-CoV-2 experiment was performed in triplicate biological and technical replications.
Pseudoviruses experiments
To verify the activity of the tested compound on PRE infection step, the assay with pseudoviruses was conducted. Briefly, HeLa[ACE2 cells were seeded in 96-well plates, cultured for 24 h at 37 °C with 5% CO2, and pre-incubated with tested compounds (60 µM) for 30 min at 37 °C and then transduced with pseudoviruses harboring VSV-G or S-SARS-CoV-2 proteins or lacking the fusion protein (ΔEnv) in the presence of polybrene (4 mg/ml, Sigma-Aldrich, Poland). After 4 h of incubation at 37 °C, unbound virions were removed by three washes with PBS, and cells were further cultured for 72 h at 37 °C with 5% CO2. Cells were lysed in Bright-Glo luciferase assay buffer (Promega, Poland) and transferred onto white 96-well plates. Luminescence levels were measured on SpectraMax iD5 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA, USA).
Isolation of nucleic acids, reverse transcription, and quantitative PCR
A viral DNA/RNA kit (A&A Biotechnology, Gdansk, Poland) was used for nucleic acid isolation from cell culture supernatants. RNA was isolated according to the manufacturer’s instructions. Viral RNA was quantified using quantitative PCR coupled with reverse transcription (RT-qPCR) (GoTaq Probe 1-Step RT-qPCR System, Promega, Poland) using a CFX96 Touch real-time PCR detection system (Bio-Rad, Munich, Germany). The reaction was carried out in the presence of the probes and primers (Fwd: CAC ATT GGC ACC CGC AAT C; Rev: GAG GAA CGA GAA GAG GCT TG; probe: 6FAM-ACT TCC TCA AGG AAC AAC ATT GCC A-BHQ-1). The heating scheme was as follows: 15 min at 45 °C and 2 min at 95 ℃, followed by 40 cycles of 15 s at 95 ℃ and 1 min at either 58 ℃ or 60 ℃. In order to assess the copy number of the N gene, standards were prepared. The PCR product was amplified and cloned into pTZ57R/T plasmids using an InsTAclone PCR cloning kit (Thermo Scientific). The resulting plasmid was linearized, and its concentration was assessed using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA); the number of copies was deducted based on the Avogadro constant. Eight 10-fold serial dilutions were used as a qPCR template to develop a standard curve.
Statistical analyses
The results are expressed as mean ± standard error of the mean (SEM). The statistical significance of the data presented in the manuscript was assessed with the non-parametric Kruskal–Wallis test, and P values below 0.05 were considered significant unless stated otherwise. Statistical analysis was performed using GraphPad Prism 9 (GraphPad Software Inc., San Diego, CA). For the determination of the half-maximal inhibitory concentration (IC50), a dose–response curve fit was performed by using a nonlinear regression model.
Data availability
Data is provided within the manuscript and supplementary information files. Further details are available under reasonable request to the corresponding authors: Ying Lei, Krzysztof Pyrc and Claudio Santi.
References
Peiris, J. S. M., Guan, Y. & Yuen, K. Y. severe acute respiratory syndrome. Nat. Med. 10, S88–S97 (2004).
Memish, Z. A., Perlman, S., Van Kerkhove, M. D. & Zumla, A. Middle east respiratory syndrome. Lancet. 395, 1063–1077 (2020).
Cannalire, R. et al. SARS-CoV-2 entry inhibitors: small molecules and peptides targeting virus or host cells. Int. J. Mol. Sci. 21, 5707 (2020).
Jin, Z. et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. bioRxiv. (2020). https://doi.org/10.1101/2020.02.26.964882
Nadeem, M. S. et al. Origin, potential therapeutic targets and treatment for Coronavirus Disease (COVID-19). Pathogens. 9, 307 (2020).
dos Santos Nascimento, I. J., da Silva-Júnior, E. F. & de Aquino, T. M. Molecular modeling targeting transmembrane serine protease 2 (TMPRSS2) as an Alternative Drug Target against coronaviruses. Curr. Drug Targets. 23, 240–259 (2022).
Perez-Miller, S. et al. Novel compounds Targeting Neuropilin receptor 1 with potential to interfere with SARS-CoV-2 Virus Entry. ACS Chem. Neurosci. 12, 1299–1312 (2021).
Harrison, C. Drug researchers pursue new lines of attack against COVID-19. Nat. Biotechnol. 38, 659–662 (2020).
Ghahremanpour, M. M. et al. Identification of 14 known drugs as inhibitors of the Main protease of SARS-CoV-2. ACS Med. Chem. Lett. 11, 2526–2533 (2020).
Owen, D. R. et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Sci. (1979). 374, 1586–1593 (2021).
Chen, X. et al. Preclinical evaluation of the SARS-CoV-2 Mpro inhibitor RAY1216 shows improved pharmacokinetics compared with nirmatrelvir. Nat. Microbiol. 9, 1075–1088 (2024).
Saar, K. L. et al. Turning high-throughput structural biology into predictive inhibitor design. Proc. Natl. Acad. Sci. 120, (2023).
Parnham, M. J. & Sies, H. The early research and development of ebselen. Biochem. Pharmacol. 86, 1248–1253 (2013).
Santi, C., Scimmi, C. & Sancineto, L. Ebselen and analogues: pharmacological properties and synthetic strategies for their Preparation. Molecules. 26, 4230 (2021).
Jin, Z. et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 582, 289–293 (2020).
Weglarz-Tomczak, E. et al. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep. 11, 3640 (2021).
Zmudzinski, M. et al. Ebselen derivatives inhibit SARS-CoV-2 replication by inhibition of its essential proteins: PLpro and Mpro proteases, and nsp14 guanine N7-methyltransferase. Sci. Rep. 13, 9161 (2023).
Menéndez, C. A., Byléhn, F., Perez-Lemus, G. R., Alvarado, W. & de Pablo, J. J. Molecular characterization of Ebselen binding activity to SARS-CoV-2 main protease. Sci. Adv. Eabd. 3045. https://doi.org/10.1126/sciadv.abd0345 (2020).
Węglarz-Tomczak, E., Tomczak, J., Talma, M. & Brul, S. Ebselen as a highly active inhibitor of PL pro CoV2. bioRxiv. https://doi.org/10.1101/2020.05.17.100768 (2020).
Mukherjee, S. et al. Ebselen inhibits Hepatitis C Virus NS3 helicase binding to nucleic acid and prevents viral replication. ACS Chem. Biol. 9, 2393–2403 (2014).
Chiou, J. et al. Ebselen as a potent covalent inhibitor of New Delhi metallo-β-lactamase (NDM-1). Chem. Commun. 51, 9543–9546 (2015).
Favrot, L. et al. Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen. Nat. Commun. 4, 2748 (2013).
Thenin-Houssier, S. et al. Ebselen, a small-molecule capsid inhibitor of HIV-1 replication. Antimicrob. Agents Chemother. 60, 2195–2208 (2016).
Leroux, F. et al. Identification of ebselen as a potent inhibitor of insulin degrading enzyme by a drug repurposing screening. Eur. J. Med. Chem. 179, 557–566 (2019).
Lenardão, E. J., Santi, C. & Sancineto, L. New frontiers in organoselenium compounds (Springer International Publishing, 2018). https://doi.org/10.1007/978-3-319-92405-2
Sies, H. & Parnham, M. J. Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections. Free Radic Biol. Med. 156, 107–112 (2020).
Amporndanai, K. et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 12, 3061 (2021).
Sahoo, P. et al. Detailed insights into the inhibitory mechanism of New Ebselen derivatives against main protease (M pro) of severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2). ACS Pharmacol. Transl Sci. https://doi.org/10.1021/acsptsci.2c00203 (2022).
Huff, S. et al. Discovery and mechanism of SARS-CoV-2 main protease inhibitors. J. Med. Chem. Acs Jmedchem. https://doi.org/10.1021/acs.jmedchem.1c00566 (2021).
Qiao, Z. et al. The Mpro structure-based modifications of ebselen derivatives for improved antiviral activity against SARS-CoV-2 virus. Bioorg. Chem. 117, 105455 (2021).
Abian, O. et al. Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening. Int. J. Biol. Macromol. 164, 1693–1703 (2020).
Mangiavacchi, F. et al. Seleno-functionalization of Quercetin improves the non-covalent inhibition of Mpro and its antiviral activity in cells against SARS-CoV-2. Int. J. Mol. Sci. 22, 7048 (2021).
Gomes, L. S. et al. Ecofriendly aminochalcogenation of alkenes: a green alternative to obtain compounds with potential anti-SARS-CoV-2 activity. New J. Chem. https://doi.org/10.1039/D2NJ06218F (2023).
Omage, F. B. et al. Diphenyl diselenide and SARS-CoV-2: in silico exploration of the mechanisms of inhibition of main protease (Mpro) and papain-like protease (PLpro). J. Chem. Inf. Model. 63, 2226–2239 (2023).
Krasowska, D. et al. Ultrasound-assisted synthesis of alkali metals diselenides (M2Se2) and their application for the gram-scale preparation of 2,2’-diselenobis(benzoic acid). Arkivoc 24–37 (2019).
Begini, F. et al. Continuous flow synthesis of 2,2′-diselenobis(benzoic acid) and derivatives. React. Chem. Eng. 5, 641–644 (2020).
Sancineto, L. et al. Design and synthesis of DiselenoBisBenzamides (DISeBAs) as nucleocapsid protein 7 (NCp7) inhibitors with anti-HIV activity. J. Med. Chem. 58, 9601–9614 (2015).
Nascimento, V. et al. Fast and easy conversion of Ortho amidoaryldiselenides into the corresponding ebselen-like derivatives driven by theoretical investigations. New J. Chem. 44, 9444–9451 (2020).
Pacuła, A. J. et al. New Chiral Ebselen analogues with antioxidant and cytotoxic potential. Molecules. 22, 492 (2017).
Obieziurska, M. et al. Bioselectivity Induced by Chirality of New Terpenyl Organoselenium compounds. Materials. 12, e3579 (2019).
Rizzuti, B. et al. Sub-micromolar inhibition of SARS-CoV-2 3CLpro by natural compounds. Pharmaceuticals. 14, 892 (2021).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 61, 3891–3898 (2021).
Santos Nascimento, I. J., de dos, Aquino, T. M. & Silva-Júnior, E. F. Da. Repurposing FDA-approved drugs targeting SARS-CoV2 3CLpro: a study by applying virtual screening, molecular dynamics, MM-PBSA calculations and covalent docking. Lett. Drug Des. Discov. 19, 637–653 (2022).
Parise, A., Romeo, I., Russo, N. & Marino, T. The Se–S bond formation in the covalent inhibition mechanism of SARS-CoV-2 main protease by Ebselen-like inhibitors: a computational study. Int. J. Mol. Sci. 22, 9792 (2021).
Madabeni, A., Nogara, P. A., Omage, F. B., Rocha, J. B. T. & Orian, L. Mechanistic insight into SARS-CoV-2 Mpro inhibition by Organoselenides: the Ebselen Case Study. Appl. Sci. 11, 6291 (2021).
Nogueira, C. W., Barbosa, N. V. & Rocha, J. B. T. Toxicology and pharmacology of synthetic organoselenium compounds: an update. Arch. Toxicol. 95, 1179–1226 (2021).
Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 263, 17205–17208 (1988).
Silva, M. S. et al. Selenium-NMR spectroscopy in Organic synthesis: from structural characterization toward New investigations. Asian J. Org. Chem. 10, 91–128 (2021).
Sands, K. N., Burman, A. L., Ansah-Asamoah, E. & Back, T. G. Chemistry related to the Catalytic cycle of the antioxidant Ebselen. Molecules. 28, 3732 (2023).
Ma, C. et al. Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin are nonspecific promiscuous SARS-CoV-2 main protease inhibitors. ACS Pharmacol. Transl Sci. 3, 1265–1277 (2020). Ebselen.
Rieder, G. S. et al. Computational analysis of the interactions between Ebselen and derivatives with the active site of the main protease from SARS-CoV-2. Comput. Biol. Chem. 107, 107956 (2023).
Ma, C., Tan, H., Choza, J., Wang, Y. & Wang, J. Validation and invalidation of SARS-CoV-2 main protease inhibitors using the Flip-GFP and protease-glo luciferase assays. Acta Pharm. Sin B. 12, 1636–1651 (2022).
Tan, H., Ma, C. & Wang, J. Invalidation of dieckol and 1,2,3,4,6-pentagalloylglucose (PGG) as SARS-CoV-2 main protease inhibitors and the discovery of PGG as a papain-like protease inhibitor. Med. Chem. Res. 31, 1147–1153 (2022).
Heilmann, E. et al. A VSV-based assay quantifies coronavirus Mpro/3CLpro/Nsp5 main protease activity and chemical inhibition. Commun. Biol. 5, 391 (2022).
Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 1-20 (2010).
Glendening, E. D. et al. NBO 7.0. Preprint at (2018).
Rizzuti, B. et al. Rutin is a low micromolar inhibitor of SARS-CoV-2 main protease 3CLpro: implications for drug design of Quercetin Analogs. Biomedicines. 9, 375 (2021).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Sci. (1979). 368, 409–412 (2020).
Hanwell, M. D. et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 17 (2012).
Santofimia-Castaño, P. et al. Intrinsically disordered chromatin protein NUPR1 binds to the C-terminal region of polycomb RING1B. Proc. Natl. Acad. Sci. 114, E6332–E6341 (2017).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph Model. 25, 247–260 (2006).
Frisch, M. J. et al. Gaussian 16, Revision C.01,. Preprint at (2016).
Fellowes, T. & White, J. M. Simulating chalcogen bonding using molecular mechanics: a pseudoatom approach to model ebselen. J. Mol. Model. 28, 66 (2022).
Maier, J. A. et al. ff14SB: improving the Accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Berendsen, H. J. C., van der Spoel, D. & van Drunen, R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 (1995).
Lindahl, E., Hess, B. & van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model 7, 306–317 (2001).
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: algorithms for highly efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 29, 845–854 (2013).
Markidis, S. & Laure, E. Solving software challenges for exascalevol. 8759 (Springer International Publishing, 2015).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX . 1–2, 19–25 (2015).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 1-8 (2007).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Miller, B. R. et al. MMPBSA.py: an efficient program for end-state Free Energy calculations. J. Chem. Theory Comput. 8, 3314–3321 (2012).
Reed, L. J. & Muench, H. A simple method of estimating 50% endpoints. Am. J. Epidemiol. 27, 493–497 (1938).
Acknowledgements
This work was undertaken under the umbrella of the Selenium Sulfur Redox & Catalysis Network (SeSRedCat).
Funding
(CS) This research was supported by Department of Pharmaceutical Sciences with the project DELPHI “Dipartimenti di Eccellenza 2018–2022” and by University of Perugia with project “Ricerca di Base”.
(KP) This work was supported by the DURABLE project, co-funded by the European Union, under the EU4Health Programme (EU4H) (https://health.ec.europa.eu/funding/eu4health-programme-2021-2027-vision-healthiereuropean-union_en#work-programmes), and ERAnet ICRAD—project Musecov: Multiscale Eco-evolution of Coronaviruses: from surveillance toward emergence prediction (https://www.era-learn.eu/network-information/networks/icrad/1st-icrad-call-2019/multiscale-eco-evolution-of-coronaviruses-from-surveillance-toward-emergence-prediction).
Author information
Authors and Affiliations
Contributions
Conceptualization, JK, JS, YL, KP, CS; methodology, AD, JK, JS, YL, CS; formal analysis, LS, FM, AD, CSc, VC, JK, GC, BR, AK-P, JL, MB, LO; investigation, LS, FM, AD, AJP, MOF, CSc, VC, JK, YZ, GC, VN, BR, AK-P, YL, MB, LO; data curation, LS, JK, GC, YL, KP, MB, LO; resources, JK, JS, YL, KP, CS; software, GC, BR, MB, LO; writing original draft preparation, LS, JK, GC, CS; writing review and editing, LS, AD, JK, GC, BR, AK-P, HY, YL, KP, MB, LO, CS;
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Institutional review board statement
Not applicable.
Informed consent
Statement: Not applicable.
Patient and public involvement
Patients or human data were not involved in this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sancineto, L., Mangiavacchi, F., Dabrowska, A. et al. New insights in the mechanism of the SARS-CoV-2 Mpro inhibition by benzisoselenazolones and diselenides. Sci Rep 14, 24751 (2024). https://doi.org/10.1038/s41598-024-75519-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-75519-6
