
Pseudo-Dipeptide Bearing α,α

 ,
,  , ,
, ,  ,
,  , , and
, , and
Abstract
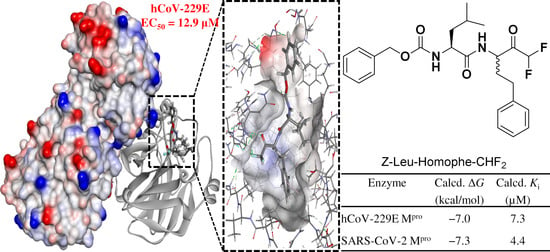
:
1. Introduction
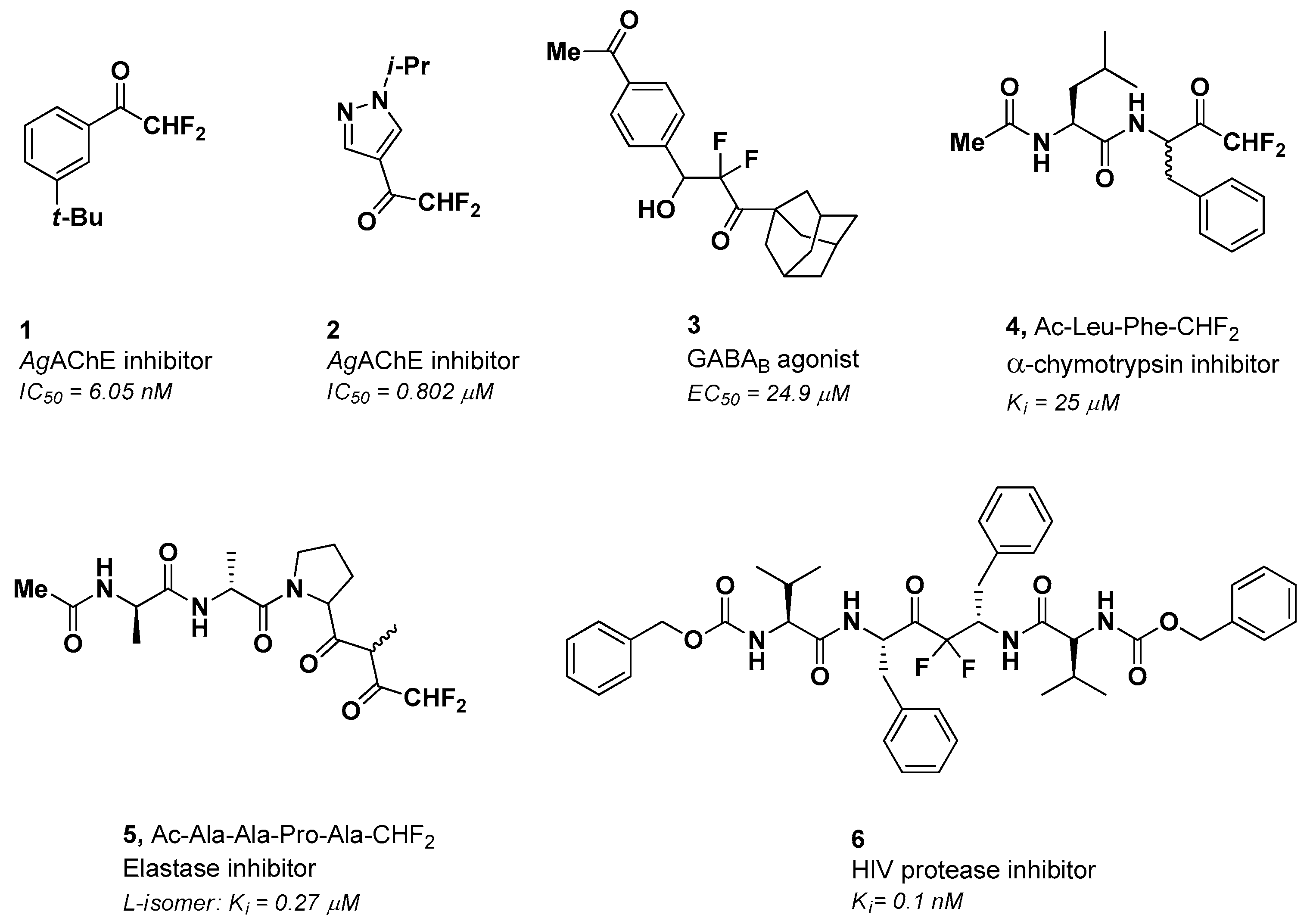
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
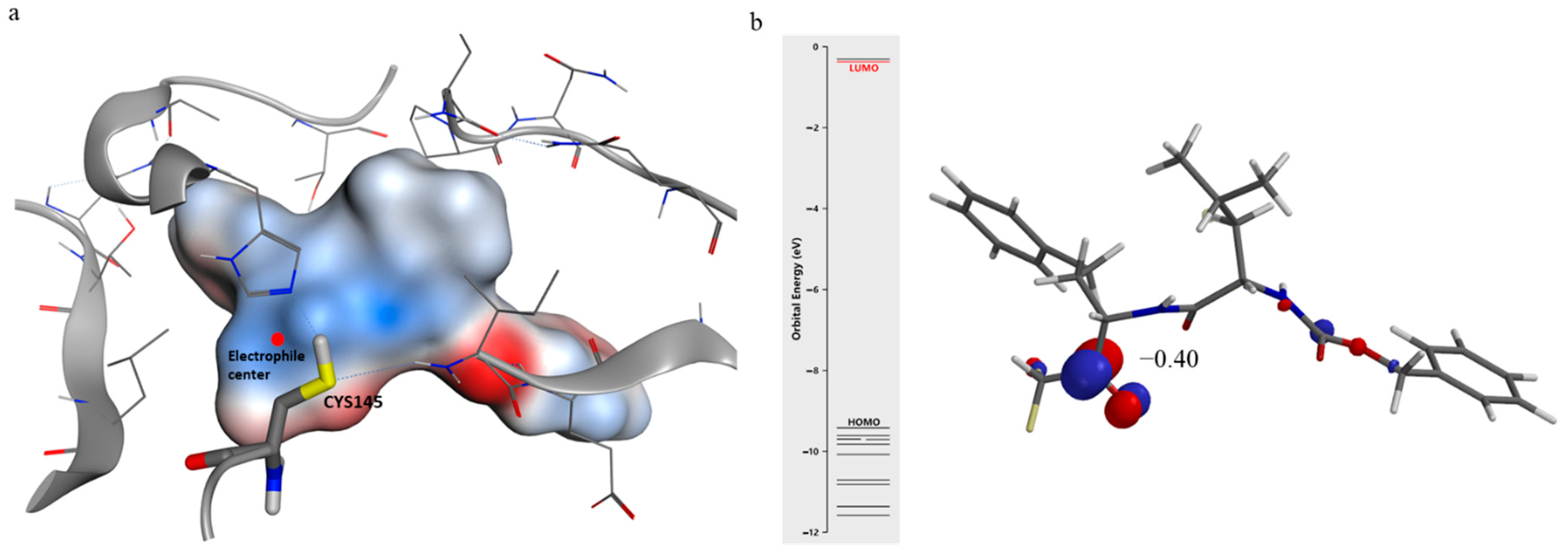
2.3. Computational
3. Materials and Methods
3.1. Chemistry
3.1.1. General Experimental Information
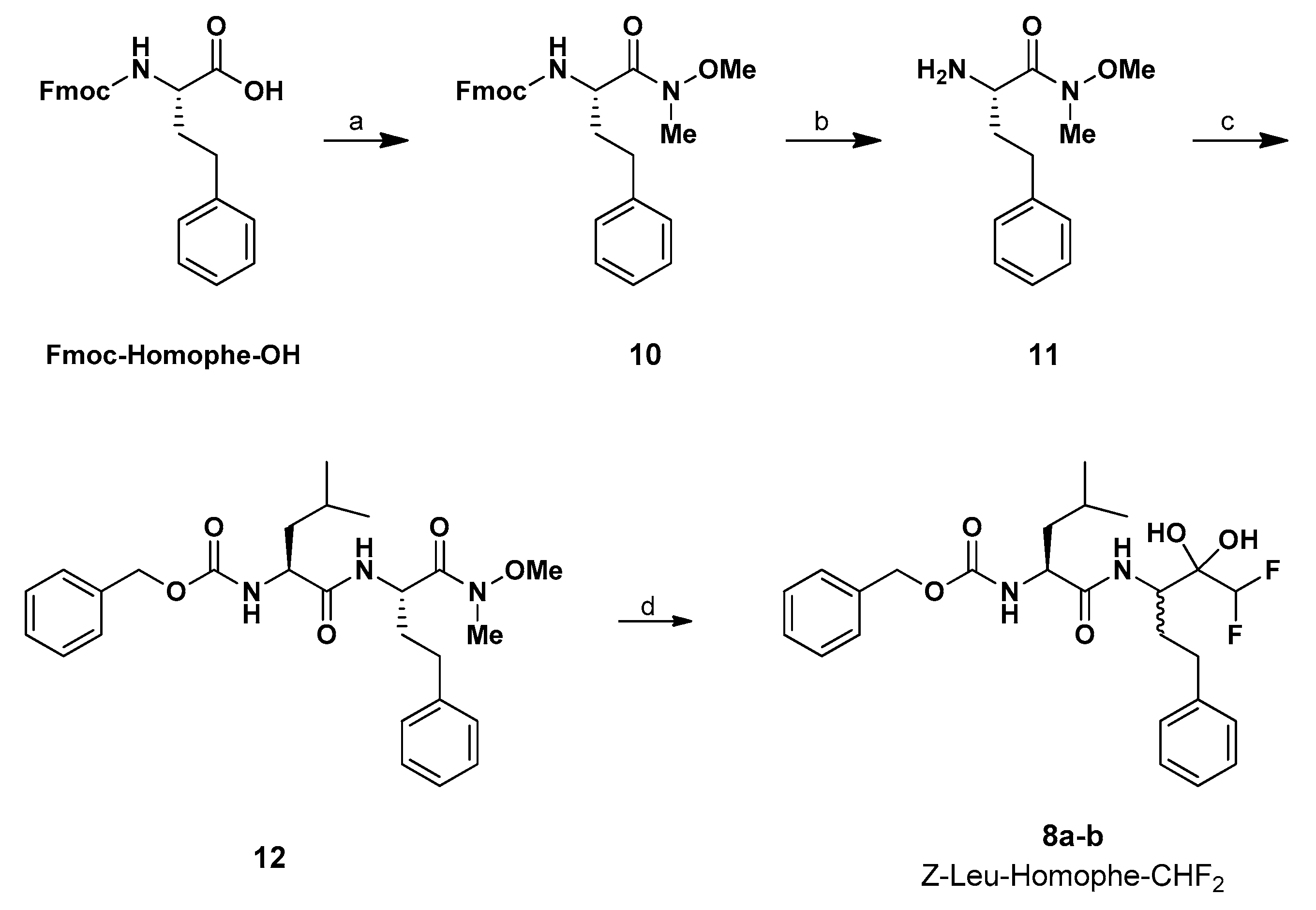
3.1.2. Synthesis of Fmoc-d-Homophe Weinreb Amide (10)
3.1.3. Synthesis of NH2-Homophe Weinreb Amide (11)
3.1.4. Synthesis of Z-Leu-Homophe Weinreb Amide (12)
3.1.5. Synthesis of Z-Leu-Homophe-CHF2 (8a,b)
3.2. Biological Assays
3.2.1. Cells and Viruses
3.2.2. Cytotoxicity Assay
3.2.3. Antiviral Assay
3.3. Computational
3.3.1. Molecular Docking
3.3.2. Molecular Optimization
3.3.3. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peeri, N.C.; Shrestha, N.; Rahman, M.S.; Zaki, R.; Tan, Z.; Bibi, S.; Baghbanzadeh, M.; Aghamohammadi, N.; Zhang, W.; Haque, U. The SARS, MERS and novel coronavirus (COVID-19) epidemics, the newest and biggest global health threats: What lessons have we learned? Int. J. Epidemiol. 2020, 49, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phanuphak, N.; Gulick, R.M. HIV treatment and prevention 2019: Current standards of care. Curr. Opin. HIV AIDS 2020, 15, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Sandmann, L.; Schulte, B.; Manns, M.P.; Maasoumy, B. Treatment of chronic hepatitis C: Efficacy, side effects and complications. Visc. Med. 2019, 35, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.D.; Urbanowicz, R.A.; Tarr, A.W.; Ball, J.K. Hepatitis C virus vaccine: Challenges and prospects. Vaccines 2020, 8, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crimi, S.; Fiorillo, L.; Bianchi, A.; D’Amico, C.; Amoroso, G.; Gorassini, F.; Mastroieni, R.; Marino, S.; Scoglio, C.; Catalano, F. Herpes virus, oral clinical signs and QoL: Systematic review of recent data. Viruses 2019, 11, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agency, E.M. Summary on Compassionate Use Remdesivir Gilead; European Medicines Agency: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Sun, D. Remdesivir for treatment of COVID-19: Combination of pulmonary and IV administration may offer additional benefit. AAPS J. 2020, 22, 77. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef] [Green Version]
- De Luca, L.; Ferro, S.; Buemi, M.R.; Monforte, A.M.; Gitto, R.; Schirmeister, T.; Maes, L.; Rescifina, A.; Micale, N. Discovery of benzimidazole-based Leishmania mexicana cysteine protease CPB 2.8 Δ CTE inhibitors as potential therapeutics for leishmaniasis. Chem. Biol. Drug Des. 2018, 92, 1585–1596. [Google Scholar] [CrossRef]
- Massai, L.; Messori, L.; Micale, N.; Schirmeister, T.; Maes, L.; Fregona, D.; Cinellu, M.A.; Gabbiani, C. Gold compounds as cysteine protease inhibitors: Perspectives for pharmaceutical application as antiparasitic agents. BioMetals 2017, 30, 313–320. [Google Scholar] [CrossRef]
- Scala, A.; Micale, N.; Piperno, A.; Rescifina, A.; Schirmeister, T.; Kesselring, J.; Grassi, G. Targeting of the leishmania Mexicana cysteine protease CPB2. 8ΔCTE by decorated fused benzo [b] thiophene scaffold. RSC Adv. 2016, 6, 30628–30635. [Google Scholar] [CrossRef] [Green Version]
- Scala, A.; Rescifina, A.; Micale, N.; Piperno, A.; Schirmeister, T.; Maes, L.; Grassi, G. Ensemble-based ADME–Tox profiling and virtual screening for the discovery of new inhibitors of the Leishmania mexicana cysteine protease CPB2. 8ΔCTE. Chem. Biol. Drug Des. 2018, 91, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Micale, N. Peptidyl fluoromethyl ketones and their applications in medicinal chemistry. Molecules 2020, 25, 4031. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Dobson, L.S.; Pattison, G. Rh-Catalyzed arylation of fluorinated ketones with arylboronic acids. Chem. Commun. 2016, 52, 11116–11119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linderman, R.J.; Jamois, E.A. A semi-empirical and ab-initio analysis of fluoroketones as reactive electrophiles. J. Fluor. Chem. 1991, 53, 79–91. [Google Scholar] [CrossRef]
- Pattison, G. Conformational preferences of α-fluoroketones may influence their reactivity. Beilstein J. Org. Chem. 2017, 13, 2915–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattison, G. Methods for the Synthesis of α, α-Difluoroketones. Eur. J. Org. Chem. 2018, 2018, 3520–3540. [Google Scholar] [CrossRef]
- Malquin, N.; Rahgoshay, K.; Lensen, N.; Chaume, G.; Miclet, E.; Brigaud, T. CF2H as a hydrogen bond donor group for the fine tuning of peptide bond geometry with difluoromethylated pseudoprolines. Chem. Commun. 2019, 55, 12487–12490. [Google Scholar] [CrossRef]
- Bordwell, F.G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Erickson, J.A.; McLoughlin, J.I. Hydrogen bond donor properties of the difluoromethyl group. J. Org. Chem. 1995, 60, 1626–1631. [Google Scholar] [CrossRef]
- Sessler, C.D.; Rahm, M.; Becker, S.; Goldberg, J.M.; Wang, F.; Lippard, S.J. CF2H, a hydrogen bond donor. J. Am. Chem. Soc. 2017, 139, 9325–9332. [Google Scholar] [CrossRef] [PubMed]
- Camerino, E.; Wong, D.M.; Tong, F.; Körber, F.; Gross, A.D.; Islam, R.; Viayna, E.; Mutunga, J.M.; Li, J.; Totrov, M.M. Difluoromethyl ketones: Potent inhibitors of wild type and carbamate-insensitive G119S mutant Anopheles gambiae acetylcholinesterase. Bioorg. Med. Chem. Lett. 2015, 25, 4405–4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowaileh, M.F.; Salyer, A.E.; Roy, K.K.; John, J.P.; Woods, J.R.; Doerksen, R.J.; Hockerman, G.H.; Colby, D.A. Agonists of the γ-aminobutyric acid type B (GABAB) receptor derived from β-hydroxy and β-amino difluoromethyl ketones. Bioorg. Med. Chem. Lett. 2018, 28, 2697–2700. [Google Scholar] [CrossRef] [PubMed]
- Govardhan, C.P.; Abeles, R.H. Structure-activity studies of fluoroketone inhibitors of α-lytic protease and human leukocyte elastase. Arch. Biochem. Biophysc. 1990, 280, 137–146. [Google Scholar] [CrossRef]
- Imperiali, B.; Abeles, R.H. Inhibition of serine proteases by peptidyl fluoromethyl ketones. Biochemistry 1986, 25, 3760–3767. [Google Scholar] [CrossRef] [PubMed]
- Sham, H.L.; Wideburg, N.E.; Spanton, S.G.; Kohlbrenner, W.E.; Betebenner, D.A.; Kempf, D.J.; Norbeck, D.W.; Plattner, J.J.; Erickson, J.W. Synthesis of (2S, 5S, 4R)-2, 5-diamino-3, 3-difluoro-1, 6-diphenylhydroxyhexane: The core unit of a potent HIV proteinase inhibitor. J. Chem. Soc. Chem. Commun. 1991, 110–112. [Google Scholar] [CrossRef]
- Kelly, C.B.; Mercadante, M.A.; Leadbeater, N.E. Trifluoromethyl ketones: Properties, preparation, and application. Chem. Commun. 2013, 49, 11133–11148. [Google Scholar] [CrossRef]
- Shao, Y.-M.; Yang, W.-B.; Kuo, T.-H.; Tsai, K.-C.; Lin, C.-H.; Yang, A.-S.; Liang, P.-H.; Wong, C.-H. Design, synthesis, and evaluation of trifluoromethyl ketones as inhibitors of SARS-CoV 3CL protease. Bioorg. Med. Chem. 2008, 16, 4652–4660. [Google Scholar] [CrossRef]
- Scala, A.; Piperno, A.; Micale, N.; Christ, F.; Debyser, Z. Synthesis and anti-HIV profile of a novel tetrahydroindazolylbenzamide derivative obtained by oxazolone chemistry. ACS Med. Chem. Lett. 2018, 10, 398–401. [Google Scholar] [CrossRef] [Green Version]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: A virtual screening and molecular modeling study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef] [Green Version]
- Piperno, A.; Cordaro, M.; Scala, A.; Iannazzo, D. Recent highlights in the synthesis of anti-HCV ribonucleosides. Curr. Med. Chem. 2014, 21, 1843–1860. [Google Scholar] [CrossRef] [PubMed]
- Pace, V.; Castoldi, L.; Mamuye, A.D.; Langer, T.; Holzer, W. Catalysis, chemoselective addition of halomethyllithiums to functionalized isatins: A straightforward access to spiro-epoxyoxindoles. Adv. Synth. Catal. 2016, 358, 172–177. [Google Scholar] [CrossRef]
- Pace, V.; Castoldi, L.; Holzer, W. Chemoselective additions of chloromethyllithium carbenoid to cyclic enones: A direct access to chloromethyl allylic alcohols. Adv. Synth. Catal. 2014, 356, 1761–1766. [Google Scholar] [CrossRef]
- Pace, V.; Castoldi, L.; Mamuye, A.D.; Holzer, W.J.S. Homologation of isocyanates with lithium carbenoids: A straightforward access to α-halomethyl-and α, α-dihalomethylamides. Synthesis 2014, 46, 2897–2909. [Google Scholar] [CrossRef]
- Pace, V.; Castoldi, L.; Mazzeo, E.; Rui, M.; Langer, T.; Holzer, W. Efficient access to all-carbon quaternary and tertiary alpha-functionalized homoallyl-type aldehydes from ketones. Angew. Chem. Int. Ed. Engl. 2017, 56, 12677–12682. [Google Scholar] [CrossRef]
- Pace, V.; Castoldi, L.; Monticelli, S.; Rui, M.; Collina, S.J.S. New perspectives in lithium carbenoid mediated homologations. Synlett 2017, 28, 879–888. [Google Scholar] [CrossRef]
- Pace, V.; Holzer, W.; Verniest, G.; Alcantara, A.R.; De Kimpe, N. Chemoselective synthesis of N-substituted α-amino-α′-chloro ketones via chloromethylation of glycine-derived weinreb amides. Adv. Synth. Catal. 2013, 355, 919–926. [Google Scholar] [CrossRef]
- Pace, V.; Murgia, I.; Westermayer, S.; Langer, T.; Holzer, W. Highly efficient synthesis of functionalized alpha-oxyketones via Weinreb amides homologation with alpha-oxygenated organolithiums. Chem. Commun. 2016, 52, 7584–7587. [Google Scholar] [CrossRef]
- Pace, V.; Castoldi, L.; Holzer, W. Synthesis of α, β-unsaturated α’-haloketones through the chemoselective addition of halomethyllithiums to weinreb amides. J. Org. Chem. 2013, 78, 7764–7770. [Google Scholar] [CrossRef]
- Pace, V.; Castoldi, L.; Holzer, W. Addition of lithium carbenoids to isocyanates: A direct access to synthetically useful N-substituted 2-haloacetamides. Chem. Commun. 2013, 49, 8383–8385. [Google Scholar] [CrossRef]
- Miele, M.; D’Orsi, R.; Sridharan, V.; Holzer, W.; Pace, V. Highly chemoselective difluoromethylative homologation of iso(thio)cyanates: Expeditious access to unprecedented alpha, alpha-difluoro(thio)amides. Chem. Commun. 2019, 55, 12960–12963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, M.; Citarella, A.; Micale, N.; Holzer, W.; Pace, V. Direct and chemoselective synthesis of tertiary difluoroketones via weinreb amide homologation with a CHF2-carbene equivalent. ACS Org. Lett. 2019, 21, 8261–8265. [Google Scholar] [CrossRef] [PubMed]
- Ielo, L.; Touqeer, S.; Roller, A.; Langer, T.; Holzer, W.; Pace, V. Telescoped, divergent, chemoselective C1 and C1-C1 homologation of imine surrogates: Access to quaternary chloro- and halomethyl-trifluoromethyl aziridines. Angew. Chem. Int. Ed. Engl. 2019, 58, 2479–2484. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, L.; Monticelli, S.; Senatore, R.; Ielo, L.; Pace, V. Homologation chemistry with nucleophilic alpha-substituted organometallic reagents: Chemocontrol, new concepts and (solved) challenges. Chem. Commun. 2018, 54, 6692–6704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, G.; Colella, M.; Monticelli, S.; Romanazzi, G.; Holzer, W.; Langer, T.; Degennaro, L.; Pace, V.; Luisi, R. Exploiting a “Beast” in carbenoid chemistry: Development of a straightforward direct nucleophilic fluoromethylation strategy. J. Am. Chem. Soc. 2017, 139, 13648–13651. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, L.; Ielo, L.; Hoyos, P.; Hernáiz, M.J.; De Luca, L.; Alcántara, A.R.; Holzer, W.; Pace, V. Merging lithium carbenoid homologation and enzymatic reduction: A combinative approach to the HIV-protease inhibitor nelfinavir. Tetrahedron 2018, 74, 2211. [Google Scholar] [CrossRef]
- Lin, G.; Liu, H.C.; Wu, F.C.; Chen, S.J. Synthesis of aryl α, α-difluoroalkyl ketones as potent inhibitors of cholesterol esterase. J. Chin. Chem. Soc. 1994, 41, 103–108. [Google Scholar] [CrossRef]
- Reiter, L.A.; Martinelli, G.J.; Reeves, L.A.; Mitchell, P.G. Difluoroketones as inhibitors of matrix metalloprotease-13. Bioorg. Med. Chem. Lett. 2000, 10, 1581–1584. [Google Scholar] [CrossRef]
- Stewart, R.; Linden, R.V.d. The acidity of some aromatic fluoro alcohols and ketones. Can. J. Chem. 1960, 38, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and sources of endemic human coronaviruses. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2018; Volume 100, pp. 163–188. [Google Scholar]
- Brown, A.J.; Wona, J.J.; Graham, R.L.; Dinnon III, K.H.; Sims, A.C.; Feng, J.Y.; Cihlarb, T.; Denison, M.R.; Baric, R.S.; Sheahan, T.P. Broad spectrum antiviral remdesivir inhibits human endemic and zoonotic deltacoronaviruses with a highly divergent RNA dependent RNA polymerase. Antivir. Res. 2019, 169, 104541. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziebuhr, J.; Heusipp, G.; Siddell, S.G. Biosynthesis, purification, and characterization of the human coronavirus 229E 3C-like proteinase. J. Virol. 1997, 71, 3992–3997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; Von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, C.; Tan, W.; Yang, K.; Yang, H. Structure of main protease from human coronavirus NL63: Insights for wide spectrum anti-coronavirus drug design. Sci. Rep. 2016, 6, 22677. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Luganini, A.; Caposio, P.; Mondini, M.; Landolfo, S.; Gribaudo, G. New cell-based indicator assays for the detection of human cytomegalovirus infection and screening of inhibitors of viral immediate-early 2 protein activity. J. Appl. Microbiol. 2008, 105, 1791–1801. [Google Scholar] [CrossRef]
- Gentile, D.; Fuochi, V.; Rescifina, A.; Furneri, P.M. New anti SARS-Cov-2 targets for quinoline derivatives chloroquine and hydroxychloroquine. Int. J. Mol. Sci. 2020, 21, 5856. [Google Scholar] [CrossRef]
- Amata, E.; Dichiara, M.; Gentile, D.; Marrazzo, A.; Turnaturi, R.; Arena, E.; La Mantia, A.; Tomasello, B.R.; Acquaviva, R.; Di Giacomo, C. Sigma receptor ligands carrying a nitric oxide donor nitrate moiety: Synthesis, in silico, and biological evaluation. ACS Med. Chem. Lett. 2020, 11, 889–894. [Google Scholar] [CrossRef]
- Floresta, G.; Patamia, V.; Gentile, D.; Molteni, F.; Santamato, A.; Rescifina, A.; Vecchio, M. Repurposing of FDA-approved drugs for treating iatrogenic botulism: A paired 3D-QSAR/docking approach. Chem. Med. Chem. 2020, 15, 256–262. [Google Scholar] [CrossRef]
- Floresta, G.; Gentile, D.; Perrini, G.; Patamia, V.; Rescifina, A. Computational tools in the discovery of FABP4 ligands: A statistical and molecular modeling approach. Mar. Drugs 2019, 17, 624. [Google Scholar] [CrossRef] [Green Version]
- Floresta, G.; Amata, E.; Gentile, D.; Romeo, G.; Marrazzo, A.; Pittalà, V.; Salerno, L.; Rescifina, A. Fourfold filtered statistical/computational approach for the identification of imidazole compounds as HO-1 inhibitors from natural products. Mar. Drugs 2019, 17, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floresta, G.; Dichiara, M.; Gentile, D.; Prezzavento, O.; Marrazzo, A.; Rescifina, A.; Amata, E. Morphing of ibogaine: A successful attempt into the search for sigma-2 receptor ligands. Int. J. Mol. Sci. 2019, 20, 488. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Prot. Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinform. 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.J. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Calcd. ΔG | Calcd. Ki (µM) |
|---|---|---|
| hCoV-229E Mpro | −7.0 | 7.3 |
| SARS-CoV-2 Mpro | −7.3 | 4.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Citarella, A.; Gentile, D.; Rescifina, A.; Piperno, A.; Mognetti, B.; Gribaudo, G.; Sciortino, M.T.; Holzer, W.; Pace, V.; Micale, N.
Pseudo-Dipeptide Bearing α,α
Citarella A, Gentile D, Rescifina A, Piperno A, Mognetti B, Gribaudo G, Sciortino MT, Holzer W, Pace V, Micale N.
Pseudo-Dipeptide Bearing α,α
Citarella, Andrea, Davide Gentile, Antonio Rescifina, Anna Piperno, Barbara Mognetti, Giorgio Gribaudo, Maria Teresa Sciortino, Wolfgang Holzer, Vittorio Pace, and Nicola Micale.
2021. "Pseudo-Dipeptide Bearing α,α