
Abstract
Coronavirus disease 2019 (COVID-19) is a rapidly growing pandemic that requires urgent therapeutic intervention. Finding potential anti COVID-19 drugs aside from approved vaccines is progressively going on. The chemically diverse natural products represent valuable sources for drug leads. In this study, we aimed to find out safe and effective COVID-19 protease inhibitors from a library of natural products which share the main nucleus/skeleton of FDA-approved drugs that were employed in COVID-19 treatment guidelines or repurposed by previous studies. Our library was subjected to virtual screening against SARS-CoV Main protease (Mpro) using Molecular Operating Environment (MOE) software. Twenty-two out of those natural candidates showed higher binding scores compared to their analogues. We repurpose these natural products including alkaloids, glucosinolates, and phenolics as potential platforms for the development of anti-SARS-CoV-2 therapeutics. This study paves the way towards discovering a lead used in the treatment of COVID-19 from natural sources and introduces phytomedicines with dual therapeutic effects against COVID-19 besides their original pharmacological effects. We recommend further in vitro evaluation of their anti-COVID-19 activity and future clinical studies.
Similar content being viewed by others

Introduction
SARS-CoV-2 is highly contagious virus, rapidly spreading and causing a global outbreak named “COVID-19” (Chen et al. 2020; Huang et al. 2020, Zhu et al., 2020 2019). Now, the coronavirus COVID-19 is affecting almost all countries around the world necessitating the rapid development of effective drugs especially with the emergence of new hot spots of COVID-19 infection within each continent in the world. Several vaccines are in the last phases of clinical trials before approval from FDA (Conte et al. 2020; Hodgson et al. 2020; Jain et al. 2020; Kaur and Gupta 2020; Lin et al. 2020; Sanchez-Felipe et al. 2020; Xia et al. 2020) and few vaccines have already been FDA approved and currently used (Knoll and Wonodi 2020; Ramasamy et al. 2020; Voysey et al. 2020; van Doremalen et al. 2020; Logunov et al. 2020; Polack et al. 2020; Folegatti et al. 2020).
Massive and fair distribution of COVID-19 vaccines represents a logistic problem challenged by the rapid emergence of new variants of COVID-19 that could escape the developed vaccines (Rubin 2021; Harvey et al. 2021), aside from increased concerns about the side effects of the approved vaccines which influence the global compliance (Wang et al. 2020, Russell and Greenwood 2020, Flanagan et al. 2020, Ledford 2021) and impart more challenges on developing potential COVID-19 antiviral drugs (Sharpe et al. 2020, Thanh et al., 2020). Pharmaceutical therapeutic strategies adapted from previous respiratory viral infections have been followed by some local guidelines, some of them have provided considerable therapeutic efficiency against COVID-19 (Rahman et al. 2020).
Virtual screening has been employed in drug discovery against emerging and fatal diseases including SARS CoV proteases (Sirois et al. 2004), hepatitis C virus RNA polymerase (Elhefnawi et al. 2012), dengue virus (Zhou et al. 2008), and ebola virus (Raj and Varadwaj 2016).
Several studies have recently employed in silico investigation of large sets of natural active constituents of plant origin using online databases in attempt to discover new hits against potential molecular targets (Shaldam et al. 2021, Al Naggar et al. 2021, Sayed et al. 2020, Owis et al. 2020, Divya et al. 2020, Rosales-Mendoza 2020, Tahir ul Qamar et al. 2020, Orhan and Senol Deniz 2020). ACE2 acts as a portal gate for the SARS-CoV-2, to enter the cell, by interacting with the viral glycoprotein spikes; this step represents a milestone for designing therapeutic interventions against COVID-19 (Behl et al. 2020). The known coronavirus 3-chymotrypsin-like protease (3CLpro), also known as Mpro, is the main protease, which is required for proteolytic maturation of the corona virus (Zhou et al. 2019; Berry et al. 2015). Targeting SARS-CoV Main protease (Mpro) enzyme will inhibit the viral maturation and enhance the host innate immune response against COVID-19 (Elmorsy et al. 2021, Shaldam et al. 2021, Jin et al. 2020, Tahir ul Qamar et al. 2020, Nguyen et al. 2012).
Kandeel and Al-Nazawi docked 487 FDA-approved drugs against Mpro and listed the top nineteen as effective candidates against COVID-19 (Kandeel and Al-Nazawi 2020). Relying on their study, clinical data from different guidelines of COVID-19 treatment in different countries, and feedback from published in vitro, in silico, or clinical studies that demonstrate effect or expected effect of repurposed drugs against COVID-19 (Kandeel and Al-Nazawi 2020; Oliveira et al. 2020; Wu et al. 2020), we determined the natural or plant derived substitute of these recently investigated repurposed drugs based on harboring a similar chemical nucleus or skeleton. Our similar chemical nucleus-based approach identified several phytochemical active agents of flavonoids, coumarins, alkaloids, terpenes, phenols, or glucosinolate origin. We decided to perform deep in silico investigation via molecular docking of these natural analogues against COVID-19 main protease to test possible activity of those naturally derived compounds compared to their chemical relatives which are already proved to have Mpro binding affinity by molecular docking. Our chemical structure-based approach identified 32 bioactive compounds of pure natural origin and reported to exhibit broad medicinal effects. These naturally existing compounds shared structural similarity (low to high) with their synthetic analogues and 14 of them displayed enhanced Mpro binding affinity greater than their analogues. Most of these chemical active constituents of plant or natural origin or their derivatives are well identified to possess wide range of pharmacological activities like anticancer, antioxidant, antihypertensive, hypoglycemic, antipyretic, antimalarial, bronchodilator, and antispasmodic which represent further advantage to treat the associated complications of COVID-19 (Nagu et al. 2021; Kashyap et al. 2021; Ghosh et al. 2021) or to provide dual therapeutic effect in patients with chronic diseases (hypertensive, diabetic, asthmatic, gouty arthritic, and hepatic) or cancer patients that have been infected with COVID-19.
Materials and methods
Molecular docking of the compounds in this study was carried out using Molecular Operating Environment 2009 (MOE) as previously described by Nagah et al. (Nagah et al. 2021). Briefly, the compounds were constructed in 3D structure, and their energies were minimized and saved to MDB file. COVID-19 main protease X-ray crystallographic structure coded as 6LU7 was downloaded together with its natural ligand (N-[(5-methylisoxazol-3-yl)carbonyl]alanyl-l-valyl-n ~ 1 ~ -((1r,2z)-4-(benzyloxy)-4-oxo-1-{[(3r)-2-oxopyrrolidin-3-yl]methyl}but-2-enyl)-l-leucinamide) from the Protein Data Bank. Hydrogens were added to the protein structure and missed connections and their types were corrected automatically. The receptor and its atoms potential were fixed. The active site of the enzyme was determined based on co-downloaded natural ligand and using site finder, dummy atoms were created to assign pocket of activity after removing natural ligand to free the pocket.
The constructed compounds’ database was docked against Mpro using the following parameters: Site of docking, dummy atoms; placement, triangle matcher; scoring, London dG with ten retains; refinement, forcefield. The resulted poses were investigated based on their energy, root-mean square deviation (rmsd), and formed interactions (bonds).
Results and discussion
Natural substitutes identification based on shared chemical nucleus
We specified highly ranked FDA-approved drugs recently repurposed against COVID-19 (Kandeel and Al-Nazawi 2020) analyzed by virtual screening and molecular docking against COVID-19 Mpro and we look through the natural library of bioactive compounds to find similar substitutes based on shared chemical nucleus or similar chemical skeleton. The identified compounds were ranked to possess high, moderate, or low degree of structural similitude to their compared partners (Table 1).
Melatonin and artemisinin while approved and repurposed for COVID-19 in some studies, they belong to naturally or biologically produced compounds (Kandeel and Al-Nazawi 2020; Shneider et al. 2020; Zhang et al. 2020; Parlakpinar et al. 2020; El-Missiry et al. 2020; Cheong et al. 2020; Li et al. 2020; Gonzalez-Paz et al. 2020).
Docking study
Structurally analogous bioactive compounds were investigated by molecular docking against the first resolved COVID-19 crystal structure (main protease), at the same time molecular docking of FDA analogous alternatives was conducted in the same way against the same target. Results of interaction energies with Mpro pocket of COVID-19 virus and receptor amino acids involved in these interactions were extracted from molecular simulation analysis and various bioactive compounds were compared against their structural FDA-approved analogues according to binding stability of the formed complex denoted by interaction energies with Mpro (Supplemental 1, Table 2). Most natural substitutes that exhibit high resemblance to FDA chemical analogues show motivating binding scores that may attribute ligand substrate interaction to functional groups or chemical skeleton shared between the FDA and the natural analogues. Considerable number (14) of the investigated phytomedicines exhibited higher affinity and stable binding to Mpro enzyme 1.34 to 2.51 folds more than their FDA alternatives (Table 2, Supplemental 2).
Binding simulation of investigated compounds
By analyzing the substrate ligand binding mode of top ranked drugs, hydrogen bonding and hydrophobic interactions were the leading drive for binding. Generally, compounds that interact with several amino acids and side chains of the docked polypeptide like 3-indolylmethyl-glucosinolate are more able to form hydrogen bonds between free hydroxyl groups in the backbone and the interacting amino acids of the binding pocket or between hydroxyl/amino groups in amino acids and polar atoms from the chemical compound backbone. In addition to hydrogen bonding, hydrophobic interactions like noncovalent interactions between π systems and arene arene stacking interactions between aromatic rings in both ligands and substrates like the alkaloids ergotamine and ephedrine and the flavonoid quercetin, or aromatic rings and heterocycles like imidazole in the backbone of the alkaloid caffeine or pyrazine in the alkaloid colletotrichumine A or pyrimidine in case of nicotinic acid.
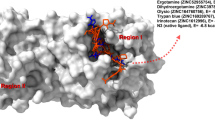
The analysis of stable protein–ligand architecture and the involvement of a specific amino acid in the ligand binding represent valuable aspects to identify and discriminate the hot-spots. From the binding simulation of tested compounds, about 15 amino acids dispersed in the Mpro binding pocket are involved in the ligand binding, most of them associate to tested compounds by hydrogen bond formation, His41 is observed to be crucial for arene arene stacking interaction, and the residues Glu166, Gly143, His41, His163, His164, Ser144, and Thr26 are the common key residues that are observed from most protein–ligand interactions.
High score alkaloids
Alkaloids that share analogous chemical nucleus like indole alkaloids reserpine from rauwolfia, vincristine and vinblastine of vinca, ergotamine and ergometrine alkaloids from ergot, terpenoid alkaloid paclitaxel of taxol, piperine from pepper and colchicine from colchicum (Evans et al. 2009) were among the top score candidates. Hydrogen bonding is mostly involved in alkaloid binding simulation to the Mpro pocket, and amino acid residues Gly143, Glu166, Gln189, and His164 of the docked polypeptide are mostly involved in the interaction with the investigated alkaloids (Figs. 1 and 2, Supplemental 2). In this study, two promising compounds reserpine and paclitaxel while exhibiting low similitude to the comparable synthetic compound unexpectedly possess high binding scores to Mpro enzyme (-26.0568 and -25.0555, respectively). Reserpine was able to dock via interaction with Gly143 of Mpro while paclitaxel exhibited two hydrogen bonds between its hydroxyl groups and Glu166 and Gln189 amino acids of Mpro (Supplemental 2). Vinblastine showed affinity to Mpro via interaction with His164 and Gln189 at a binding score of − 23.9685 (Fig. 1, Table 2). Colchicine represents another example from this group that formed hydrogen bonds with Gly143 and Gln189 via its methoxy and amido groups (Fig. 2, Table 2). Cautions should be paid for the use of such alkaloids specially the anticancer ones to avoid their side effects; however, cost benefit ratio will play a role in their use depending on the severity of the case or in case of COVID-19 cancer patients.

Amino acid interactions of COVID-19 main protease Mpro with vinblastine alkaloid in two-dimensional configuration (up) and three-dimensional configuration (down) using MOE

Amino acid interactions of COVID-19 main protease Mpro with colchicine alkaloid in two-dimensional configuration (up) and three-dimensional configuration (down) using MOE
Glucosinolates
Glucosinolates glycosides were recognized in our screening as natural structural substitutes to the carbonic anhydrase inhibitor methazolamide indicated in the treatment of increased intraocular pressure and the antiepileptic zonisamide and both drugs are repurposed against COVID-19 (Kandeel and Al-Nazawi 2020). Glucosinolates show promising binding scores and form stable complex through interaction with a cluster of amino acid residues Phe140, His164, Asn142, His163, Thr26, Leu141, and Ser144 which are assigned behind ligand substrate binding in the binding simulation (Fig. 3, supplemental 2). 3-Methylsulfinylpropyl-glucosinolate comes on the top of this group with a binding score of − 22.6547, it interacted with Phe140 and His164. On the other hand, 3-indolylmethyl-glucosinolate was the most interesting compound in this study as it stabilize itself in Mpro pocket via six different bonds with five amino acids namely Thr26, Leu141, Asn142, Ser144, and His163. It is obvious from these interactions that sulfate and glucose groups of glucosinolates are essential for activity (Fig. 3, Table 2, Supplemental 2).

Amino acid interactions of COVID-19 main protease Mpro with 3-indolylmethyl-glucosinolate in two-dimensional configuration (up) and three-dimensional configuration (down) using MOE
Flavonoids, coumarins and phenolic phytocompounds
Significant number of flavonoids and phenolic natural products came out in our screening, interestingly most of them were proposed in other studies to possess Mpro affinity and previously documented to have antiviral effect. Quercetin, caffeic acid, and o-coumaric acid were recently repurposed against COVID-19 in several studies (Colunga Biancatelli et al. 2020; Bachevski et al. 2020; Mani et al. 2020; Bhowmik et al. 2020; Elfiky 2020, Sayed et al. 2020). Flavonoids and phenolics were identified as analogues to the vasoprotective chromocarb and the phenylethanolamine octapamine used for treatment of hypotensive regulatory and circulatory disorders. Aside from these previously assigned compounds, new members like the major flavonolignan of silymarin (silybin) and khellin coumarin from Ammi visnaga show promising binding scores for COVID-19 Mpro enzyme (− 22.6614 and − 20.1254, respectively), silybin exhibited hydrogen bonds interactions through its hydroxyl groups and Thr24 and Phe140, while khellin interacted with Glu166 via one of its methoxy groups. Another interesting component from this class is salicin that exceeded the binding score of its synthetic analogue (aminosalicylate sodium) by about 1.36, it showed interactions with Phe140 and Leu141 amino acids of Mpro pocket (Fig. 4, Table 2, Supplemental 2). Like alkaloids and glucosinolates, repurposed flavonoids, coumarins, and phenolics stabilize their interaction through hydrogen bonding with amino acids including Thr26, Thr24, Phe140, Leu141, and Glu166 within the Mpro pocket (Fig. 4, supplemental 2).

Amino acid interactions of COVID-19 main protease Mpro with silybin, in two-dimensional configuration (up) and three-dimensional configuration (down) using MOE
Relation to oseltamivir (Tamiflu®)
Antiviral therapy of COVID-19 patients with lopinavir/ritonavir showed considerable efficiency according to local guidelines (Negrut et al. 2021). Oseltamivir (an antiviral agent) is approved in COVID-19 treatment protocol in many countries including Japan, China, and Egypt. Molecular docking of this compound against Mpro exhibited binding with His164 and Gln189 at a score of − 17.299. Based on in silico analysis of the docked natural products relative to oseltamivir, we found that ten compounds including khellin, silybin, 3-hydroxypropyl-glucosinolate, 3-methylsulfinylpropyl-glucosinolate, 3-benzoyloxypropyl-glucosinolate, 3-indolylmethyl-glucosinolate, vinblastine, reserpine, colchicine, and paclitaxel showed better binding scores. These results reflect the importance of natural products as drug leads for treatment of COVID-19. We highly recommend carrying out in vivo study for these compounds to confirm their importance as new and effective treatment for this COVID-19 infection and determine their effective doses.
Limitations
Our study was based on previous in silico studies of FDA-approved drugs that were virtually screened against COVID-19 Mpro; we only considered natural substitutes of these drugs that possess considerable structural similarity. A more comprehensive approach has to be considered to cover more natural candidates and phytochemicals and target other COVID-19 functional proteins in the future screening approaches.
Conclusion
We screened a library of phytochemicals based on similar analogous chemical structure or shared functional groups to FDA-approved drugs recently employed or repurposed for COVID-19 treatment. Interestingly, our screening identified some candidates of natural active agents that have been recently repurposed by virtual screening as COVID-19 Mpro inhibitors like quercetin, colchicine, piperine, and caffeic acid. Looking at compounds with high interaction energy in our screening, we identified novel bioactive compounds including antihypertensive alkaloid reserpine, potential anticancer like paclitaxel, vincristine and vinblastine alkaloids, well-known hepatoprotective silybin and other flavonoids and phenolic compounds, and more important cluster of glucosinolate glycosides naturally occurring in many pungent plants such as mustard, cabbage, broccoli, rocket, and horseradish.
Future prospects
While we propose these newly screened phytomedicines based on their binding scores as drug leads for design of potential COVID-19 Mpro inhibitors, we recommend further in vitro examination and clinical evaluation of their virtual anti COVID-19 activity.
Data availability
Not applicable.
References
Al Naggar Y, Giesy JP, Abdel-Daim MM, Javed Ansari M, Al-Kahtani SN, Yahya G (2021) Fighting against the second wave of COVID-19: can honeybee products help protect against the pandemic? Saudi Journal of Biological Sciences 28(3):1519–1527. https://doi.org/10.1016/j.sjbs.2020.12.031
Bachevski D, Damevska K, Simeonovski V, Dimova M (2020) Back to the basics: propolis and COVID-19. Dermatologic therapy:e13780. doi: https://doi.org/10.1111/dth.13780
Behl T, Kaur I, Bungau S, Kumar A, Uddin MS, Kumar C, Pal G, Sahil SK, Zengin G, Arora S (2020) The dual impact of ACE2 in COVID-19 and ironical actions in geriatrics and pediatrics with possible therapeutic solutions. Life Sci 257:118075. https://doi.org/10.1016/j.lfs.2020.118075
Berry M, Fielding B, Gamieldien J (2015) Potential broad spectrum inhibitors of the Coronavirus 3CLpro: a virtual screening and structure-based drug design study. Viruses 7(12):6642–6660. https://doi.org/10.3390/v7122963
Bhowmik D, Nandi R, Jagadeesan R, Kumar N, Prakash A, Kumar D (2020) Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infection, Genetics and Evolution : Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases 84:104451. https://doi.org/10.1016/j.meegid.2020.104451
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Xia J, Yu T, Zhang X, Zhang L (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. The Lancet 395(10223):507–513. https://doi.org/10.1016/S0140-6736(20)30211-7
Cheong DHJ, Tan DWS, Wong FWS, Tran T (2020) Anti-malarial drug, artemisinin and its derivatives for the treatment of respiratory diseases. Pharmacol Res 158:104901. https://doi.org/10.1016/j.phrs.2020.104901
Colunga Biancatelli RML, Berrill M, Catravas JD, Marik PE (2020) Quercetin and vitamin C: an experimental, synergistic therapy for the prevention and treatment of SARS-CoV-2 related disease (COVID-19). Front Immunol 11:1451. https://doi.org/10.3389/fimmu.2020.01451
Conte C, Sogni F, Affanni P, Veronesi L, Argentiero A, Esposito S (2020) Vaccines against Coronaviruses: the state of the art. Vaccines 8(2). doi: https://doi.org/10.3390/vaccines8020309
Divya M, Vijayakumar S, Chen J, Vaseeharan B, Durán-Lara EF (2020) A review of South Indian medicinal plant has the ability to combat against deadly viruses along with COVID-19? Microbial Pathogenesis:104277. doi: https://doi.org/10.1016/j.micpath.2020.104277
Elfiky AA (2020) Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics:1–10. doi: https://doi.org/10.1080/07391102.2020.1761881
Elhefnawi M, ElGamacy M, Fares M (2012) Multiple virtual screening approaches for finding new hepatitis C virus RNA-dependent RNA polymerase inhibitors: structure-based screens and molecular dynamics for the pursue of new poly pharmacological inhibitors. BMC Bioinformatics 13(Suppl 17):S5. https://doi.org/10.1186/1471-2105-13-S17-S5
El-Missiry MA, El-Missiry ZM, Othman AI (2020) Melatonin is a potential adjuvant to improve clinical outcomes in individuals with obesity and diabetes with coexistence of Covid-19. European journal of pharmacology:173329. doi: https://doi.org/10.1016/j.ejphar.2020.173329
Elmorsy, M. A., El-Baz, A. M., Mohamed, N. H., Almeer, R., Abdel-Daim, M. M., & Yahya, G. (2021) In silico screening of potent inhibitors against COVID-19 key targets from a library of FDA-approved drugs. Environmental science and pollution research international, 1–11. Advance online publication. https://doi.org/10.1007/s11356-021-16427-4
Erukainure OL, Sanni O, Islam MS (2018) Clerodendrum volubile: phenolics and applications to health. In: Watson RR, Preedy VR, Zibadi S (eds) Polyphenols. Volume 1. Mechanisms of action in human health and disease / edited by Ronald Ross Watson, Victor R. Preedy, Sherma Zibadi, Second edition. Elsevier, Amsterdam, pp 53–68
Evans WC, Evans D, Trease GE (2009) Trease and Evans pharmacognosy [electronic resource]. Saunders/Elsevier, Edinburgh, New York
Flanagan KL, Best E, Crawford NW, Giles M, Koirala A, Macartney K, Russell F, Teh BW, Wen SC (2020) Progress and pitfalls in the quest for effective SARS-CoV-2 (COVID-19) vaccines. Front Immunol 11:579250. https://doi.org/10.3389/fimmu.2020.579250
Folegatti PM, Ewer KJ, Aley PK, Angus B, Becker S, Belij-Rammerstorfer S, Bellamy D, Bibi S, Bittaye M, Clutterbuck EA, Dold C, Faust SN, Finn A, Flaxman AL, Hallis B, Heath P, Jenkin D, Lazarus R, Makinson R, Minassian AM, Pollock KM, Ramasamy M, Robinson H, Snape M, Tarrant R, Voysey M, Green C, Douglas AD, Hill AVS, Lambe T, Gilbert SC, Pollard AJ (2020) Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet (london, England) 396(10249):467–478. https://doi.org/10.1016/S0140-6736(20)31604-4
Ghosh S, Das S, Mondal R, Abdullah S, Sultana S, Singh S, Sehgal A, Behl T (2021) A review on the effect of COVID-19 in type 2 asthma and its management. Int Immunopharmacol 91:107309. https://doi.org/10.1016/j.intimp.2020.107309
Gonzalez-Paz LA, Lossada CA, Moncayo LS, Romero F, Paz JL, Vera-Villalobos J, Pérez AE, San-Blas E, Alvarado YJ (2020) Theoretical molecular docking study of the structural disruption of the viral 3CL-protease of COVID19 induced by binding of capsaicin, piperine and curcumin part 1: a comparative study with chloroquine and hydrochloroquine two antimalaric drugs
Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, Ludden C, Reeve R, Rambaut A, Peacock SJ, Robertson DL (2021) SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. https://doi.org/10.1038/s41579-021-00573-0
Hodgson SH, Mansatta K, Mallett G, Harris V, Emary KRW, Pollard AJ (2020) What defines an efficacious COVID-19 vaccine? A review of the challenges assessing the clinical efficacy of vaccines against SARS-CoV-2. The Lancet. Infectious diseases. doi: https://doi.org/10.1016/S1473-3099(20)30773-8
Hu Z, Wang J, Bi X, Zhang J, Xue Y, Yang Y, Luo Z, Yao G, Zhang Y (2014) Colletotrichumine A, a novel indole–pyrazine alkaloid with an unprecedented C16N3-type skeleton from cultures of Colletotrichum capsici. Tetrahedron Lett 55(44):6093–6095. https://doi.org/10.1016/j.tetlet.2014.09.041
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China the Lancet 395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
Jain S, Batra H, Yadav P, Chand S (2020) COVID-19 vaccines currently under preclinical and clinical studies, and associated antiviral immune response. Vaccines 8(4). doi: https://doi.org/10.3390/vaccines8040649
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582(7811):289–293. https://doi.org/10.1038/s41586-020-2223-y
Kandeel M, Al-Nazawi M (2020) Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci 251:117627. https://doi.org/10.1016/j.lfs.2020.117627
Kashyap S, Bala R, Madaan R, Behl T (2021) Uncurtaining the effect of COVID-19 in diabetes mellitus: a complex clinical management approach. Environ Sci Pollut Res Int 28(27):35429–35436. https://doi.org/10.1007/s11356-021-14480-7
Kaur SP, Gupta V (2020) COVID-19 Vaccine: a comprehensive status report. Virus Res 288:198114. https://doi.org/10.1016/j.virusres.2020.198114
Knoll MD, Wonodi C (2020) Oxford–AstraZeneca COVID-19 vaccine efficacy. The Lancet. https://doi.org/10.1016/S0140-6736(20)32623-4
Ledford H (2021) COVID vaccines and blood clots: five key questions. Nature 592(7855):495–496. https://doi.org/10.1038/d41586-021-00998-w
Li X, Zhang C, Liu L, Gu M (2020) Existing bitter medicines for fighting 2019-nCoV-associated infectious diseases. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology 34(5):6008–6016. https://doi.org/10.1096/fj.202000502
Lin D-Y, Zeng D, Mehrotra DV, Corey L, Gilbert PB (2020) Evaluating the efficacy of COVID-19 vaccines. Clinical Infectious Diseases : an Official Publication of the Infectious Diseases Society of America. https://doi.org/10.1093/cid/ciaa1863
Logunov DY, Dolzhikova IV, Tukhvatullin AI, Shcheblyakov DV (2020) Safety and efficacy of the Russian COVID-19 vaccine: more information needed - Authors’ reply. Lancet (london, England) 396(10256):e54–e55. https://doi.org/10.1016/S0140-6736(20)31970-X
Mani JS, Johnson JB, Steel JC, Broszczak DA, Neilsen PM, Walsh KB, Naiker M (2020) Natural product-derived phytochemicals as potential agents against coronaviruses: a review. Virus Res 284:197989. https://doi.org/10.1016/j.virusres.2020.197989
Mostafa I, Abbas HA, Ashour ML, Yasri A, El-Shazly AM, Wink M, Sobeh M (2020) Polyphenols from Salix tetrasperma impair virulence and inhibit quorum sensing of Pseudomonas aeruginosa. Molecules (Basel, Switzerland) 25(6). doi: https://doi.org/10.3390/molecules25061341
Mostafa I, Abd El-Aziz E, Hafez S, El-Shazly A (2013) Chemical constituents and biological activities of Galinsoga parviflora cav. (Asteraceae) from Egypt. Zeitschrift fur Naturforschung. C, Journal of biosciences 68(7–8):285–292
Mostafa I, Zhu N, Yoo M-J, Balmant KM, Misra BB, Dufresne C, Abou-Hashem M, Chen S, El-Domiaty M (2016) New nodes and edges in the glucosinolate molecular network revealed by proteomics and metabolomics of Arabidopsis myb28/29 and cyp79B2/B3 glucosinolate mutants. J Proteomics 138:1–19. https://doi.org/10.1016/j.jprot.2016.02.012
Nagah N, Mostafa I, Osman A, Dora G, El-Sayed Z, Ateya AM (2021) Bioguided isolation and in-silico analysis of Hep-G2 cytotoxic constituents from Laurus nobilis Linn. cultivated in Egypt. Egypt J Chem 64:2731–2745
Nagu P, Parashar A, Behl T, Mehta V (2021) CNS implications of COVID-19: a comprehensive review. Rev Neurosci 32(2):219–234. https://doi.org/10.1515/revneuro-2020-0070
Negrut N, Codrean A, Hodisan I, Bungau S, Tit DM, Marin R, Behl T, Banica F, Diaconu CC, Nistor-Cseppento DC, Nistor-Cseppento DC (2021) Efficiency of antiviral treatment in COVID-19. Exp Ther Med 21:648. https://doi.org/10.3892/etm.2021.10080
Nguyen TTH, Woo H-J, Kang H-K, van Nguyen D, Kim Y-M, Kim D-W, Ahn S-A, Xia Y, Kim D (2012) Flavonoid-mediated inhibition of SARS coronavirus 3C-like protease expressed in Pichia pastoris. Biotechnol Lett 34(5):831–838. https://doi.org/10.1007/s10529-011-0845-8
Oliveira OV de, Rocha GB, Paluch AS, Costa LT (2020) Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. Journal of Biomolecular Structure and Dynamics:1–10. doi: https://doi.org/10.1080/07391102.2020.1772885
Orhan IE, Senol Deniz FS (2020) Natural products as potential leads against Coronaviruses: could they be encouraging structural models against SARS-CoV-2? Nat Prod Bioprospect 10(4):171–186. https://doi.org/10.1007/s13659-020-00250-4
Owis AI, El-Hawary MS, El Amir D, Aly OM, Abdelmohsen UR, Kamel MS (2020) Molecular docking reveals the potential of Salvadora persica flavonoids to inhibit COVID-19 virus main protease. RSC Adv 10(33):19570–19575. https://doi.org/10.1039/D0RA03582C
Parlakpinar H, Polat S, Acet HA (2020) Pharmacological agents under investigation in the treatment of coronavirus disease 2019 and the importance of melatonin. Fundam Clin Pharmacol. https://doi.org/10.1111/fcp.12589
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW, Hammitt LL, Türeci Ö, Nell H, Schaefer A, Ünal S, Tresnan DB, Mather S, Dormitzer PR, Şahin U, Jansen KU, Gruber WC (2020) Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med. https://doi.org/10.1056/NEJMoa2034577
Rahman MH, Akter R, Behl T, Chowdhury M, Mohammed M, Bulbul IJ, Elshenawy SE, Kamal MA (2020) COVID-19 outbreak and emerging management through pharmaceutical therapeutic strategy. Curr Pharm Des 26(41):5224–5240. https://doi.org/10.2174/1381612826666200713174140
Raj U, Varadwaj PK (2016) Flavonoids as multi-target inhibitors for proteins associated with ebola virus: in silico discovery using virtual screening and molecular docking studies. Interdiscip Sci Comput Life Sci 8(2):132–141. https://doi.org/10.1007/s12539-015-0109-8
Ram VJ, Sethi A, Nath M, Pratap R (2019) The chemistry of heterocycles. Chemistry of six to eight membered N, O, S, P and SE heterocycles / Vishnu Ji Ram, Arun Sethi, Mahendra Nath, Ramendra Pratap. Elsevier, Amsterdam
Ramasamy MN, Minassian AM, Ewer KJ, Flaxman AL, Folegatti PM, Owens DR, Voysey M, Aley PK, Angus B, Babbage G, Belij-Rammerstorfer S, Berry L, Bibi S, Bittaye M, Cathie K, Chappell H, Charlton S, Cicconi P, Clutterbuck EA, Colin-Jones R, Dold C, Emary KRW, Fedosyuk S, Fuskova M, Gbesemete D, Green C, Hallis B, Hou MM, Jenkin D, Joe CCD, Kelly EJ, Kerridge S, Lawrie AM, Lelliott A, Lwin MN, Makinson R, Marchevsky NG, Mujadidi Y, Munro APS, Pacurar M, Plested E, Rand J, Rawlinson T, Rhead S, Robinson H, Ritchie AJ, Ross-Russell AL, Saich S, Singh N, Smith CC, Snape MD, Song R, Tarrant R, Themistocleous Y, Thomas KM, Villafana TL, Warren SC, Watson MEE, Douglas AD, Hill AVS, Lambe T, Gilbert SC, Faust SN, Pollard AJ (2020) Safety and immunogenicity of ChAdOx1 nCoV-19 vaccine administered in a prime-boost regimen in young and old adults (COV002): a single-blind, randomised, controlled, phase 2/3 trial. Lancet (London, England). doi: https://doi.org/10.1016/S0140-6736(20)32466-1
Rosales-Mendoza S (2020) Will plant-made biopharmaceuticals play a role in the fight against COVID-19? Expert Opin Biol Ther 20(6):545–548. https://doi.org/10.1080/14712598.2020.1752177
Rubin R (2021) COVID-19 Vaccines vs variants—determining how much immunity is enough. JAMA 325(13):1241. https://doi.org/10.1001/jama.2021.3370
Sanchez-Felipe L, Vercruysse T, Sharma S, Ma J, Lemmens V, van Looveren D, Javarappa MPA, Boudewijns R, Malengier-Devlies B, Liesenborghs L, Kaptein SJF, de Keyzer C, Bervoets L, Debaveye S, Rasulova M, Seldeslachts L, Li L-H, Jansen S, Yakass MB, Verstrepen BE, Böszörményi KP, Kiemenyi-Kayere G, van Driel N, Quaye O, Zhang X, Ter Horst S, Mishra N, Deboutte W, Matthijnssens J, Coelmont L, Vandermeulen C, Heylen E, Vergote V, Schols D, Wang Z, Bogers W, Kuiken T, Verschoor E, Cawthorne C, van Laere K, Opdenakker G, Velde GV, Weynand B, Teuwen DE, Matthys P, Neyts J, Jan Thibaut H, Dallmeier K (2020) A single-dose live-attenuated YF17D-vectored SARS-CoV-2 vaccine candidate. Nature. https://doi.org/10.1038/s41586-020-3035-9
Shaldam MA, Yahya G, Mohamed NH, Abdel-Daim MM, Al Naggar Y (2021) In silico screening of potent bioactive compounds from honeybee products against COVID-19 target enzymes. Environ Sci Pollut Res Int 28(30):40507–40514. https://doi.org/10.1007/s11356-021-14195-9
Sharpe HR, Gilbride C, Allen E, Belij-Rammerstorfer S, Bissett C, Ewer K, Lambe T (2020) The early landscape of COVID-19 vaccine development in the UK and rest of the world. Immunology. https://doi.org/10.1111/imm.13222
Shneider A, Kudriavtsev A, Vakhrusheva A (2020) Can melatonin reduce the severity of COVID-19 pandemic? International reviews of immunology:1–10. doi: https://doi.org/10.1080/08830185.2020.1756284
Sirois S, Wei D-Q, Du Q, Chou K-C (2004) Virtual screening for SARS-CoV protease based on KZ7088 pharmacophore points. J Chem Inf Comput Sci 44(3):1111–1122. https://doi.org/10.1021/ci034270n
Sobeh M, Mahmoud MF, Rezq S, Alsemeh AE, Sabry OM, Mostafa I, Abdelfattah MAO, El-Allem KA, El-Shazly AM, Yasri A, Wink M (2019) Salix tetrasperma Roxb. Extract alleviates neuropathic pain in rats via modulation of the NF-κB/TNF-α/NOX/iNOS pathway. Antioxidants (Basel, Switzerland) 8(10). doi: https://doi.org/10.3390/antiox8100482
Tahir ul Qamar M, Alqahtani SM, Alamri MA, Chen L-L, (2020) Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. Journal of Pharmaceutical Analysis. https://doi.org/10.1016/j.jpha.2020.03.009
Le Thanh T, Andreadakis Z, Kumar A, Gómez Román R, Tollefsen S, Saville M, Mayhew S (2020) The COVID-19 vaccine development landscape. Nat Rev Drug Discov 19(5):305–306. https://doi.org/10.1038/d41573-020-00073-5
van Doremalen N, Lambe T, Spencer A, Belij-Rammerstorfer S, Purushotham JN, Port JR, Avanzato VA, Bushmaker T, Flaxman A, Ulaszewska M, Feldmann F, Allen ER, Sharpe H, Schulz J, Holbrook M, Okumura A, Meade-White K, Pérez-Pérez L, Edwards NJ, Wright D, Bissett C, Gilbride C, Williamson BN, Rosenke R, Long D, Ishwarbhai A, Kailath R, Rose L, Morris S, Powers C, Lovaglio J, Hanley PW, Scott D, Saturday G, de Wit E, Gilbert SC, Munster VJ (2020) ChAdOx1 nCoV-19 vaccine prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature 586(7830):578–582. https://doi.org/10.1126/science.abb2507
Voysey M, Clemens SAC, Madhi SA, Weckx LY, Folegatti PM, Aley PK, Angus B, Baillie VL, Barnabas SL, Bhorat QE, Bibi S, Briner C, Cicconi P, Collins AM, Colin-Jones R, Cutland CL, Darton TC, Dheda K, Duncan CJA, Emary KRW, Ewer KJ, Fairlie L, Faust SN, Feng S, Ferreira DM, Finn A, Goodman AL, Green CM, Green CA, Heath PT, Hill C, Hill H, Hirsch I, Hodgson SHC, Izu A, Jackson S, Jenkin D, Joe CCD, Kerridge S, Koen A, Kwatra G, Lazarus R, Lawrie AM, Lelliott A, Libri V, Lillie PJ, Mallory R, Mendes AVA, Milan EP, Minassian AM, McGregor A, Morrison H, Mujadidi YF, Nana A, O'Reilly PJ, Padayachee SD, Pittella A, Plested E, Pollock KM, Ramasamy MN, Rhead S, Schwarzbold AV, Singh N, Smith A, Song R, Snape MD, Sprinz E, Sutherland RK, Tarrant R, Thomson EC, Török ME, Toshner M, Turner DPJ, Vekemans J, Villafana TL, Watson MEE, Williams CJ, Douglas AD, Hill AVS, Lambe T, Gilbert SC, Pollard AJ (2020) Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet (London, England). doi: https://doi.org/10.1016/S0140-6736(20)32661-1
Wang J, Jing R, Lai X, Zhang H, Lyu Y, Knoll MD, Fang H (2020) Acceptance of COVID-19 vaccination during the COVID-19 pandemic in China. Vaccines 8(3). doi: https://doi.org/10.3390/vaccines8030482
Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M, Chen L, Li H (2020) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B 10(5):766–788. https://doi.org/10.1016/j.apsb.2020.02.008
Xia S, Duan K, Zhang Y, Zhao D, Zhang H, Xie Z, Li X, Peng C, Zhang Y, Zhang W, Yang Y, Chen W, Gao X, You W, Wang X, Wang Z, Shi Z, Wang Y, Yang X, Zhang L, Huang L, Wang Q, Lu J, Yang Y, Guo J, Zhou W, Wan X, Wu C, Wang W, Huang S, Du J, Meng Z, an Pan, Yuan Z, Shen S, Guo W, Yang X, (2020) Effect of an inactivated vaccine against SARS-CoV-2 on safety and immunogenicity outcomes: interim analysis of 2 randomized clinical trials. JAMA 324(10):951–960. https://doi.org/10.1001/jama.2020.15543
Yuan K, Zhu Q, Lu Q, Jiang H, Zhu M, Li X, Huang G, Xu A (2020) Quercetin alleviates rheumatoid arthritis by inhibiting neutrophil inflammatory activities. J Nutr Biochem 84:108454. https://doi.org/10.1016/j.jnutbio.2020.108454
Zhang R, Wang X, Ni L, Di X, Ma B, Niu S, Liu C, Reiter RJ (2020) COVID-19: Melatonin as a potential adjuvant treatment. Life Sci 250:117583. https://doi.org/10.1016/j.lfs.2020.117583
Zhou J, Fang L, Yang Z, Xu S, Lv M, Sun Z, Chen J, Wang D, Gao J, Xiao S (2019) Identification of novel proteolytically inactive mutations in coronavirus 3C-like protease using a combined approach. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology 33(12):14575–14587. https://doi.org/10.1096/fj.201901624RR
Zhou Z, Khaliq M, Suk J-E, Patkar C, Li L, Kuhn RJ, Post CB (2008) Antiviral compounds discovered by virtual screening of small-molecule libraries against dengue virus E protein. ACS Chem Biol 3(12):765–775. https://doi.org/10.1021/cb800176t
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382(8):727–733. https://doi.org/10.1056/NEJMoa2001017
Acknowledgements
The authors would like to extend their sincere appreciation to the Researchers Supporting Project number (RSP-2021/96), King Saud University, Riyadh, Saudi Arabia. The authors would like to thank Dr. Ahmed Samy, Department of Drug Chemistry, Faculty of Pharmacy, Zagazig University, Egypt for allowing the use of MOE software.
Funding
The authors would like to extend their sincere appreciation to the Researchers Supporting Project number (RSP-2021/96), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
I.M. performed molecular docking and data analysis, participated in idea development, writing and revision of manuscript, N.H.M. participated in writing of manuscript, B.M. participated in data analysis, R.A., M.M.A.A., S.G.B., and A.M.E-S. provided reviewing and editing, and G.Y. provided the idea and wrote and revised the manuscript. All authors have read and agreed to the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Lotfi Aleya.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mostafa, I., Mohamed, N.H., Mohamed, B. et al. In-silico screening of naturally derived phytochemicals against SARS-CoV Main protease. Environ Sci Pollut Res 29, 26775–26791 (2022). https://doi.org/10.1007/s11356-021-17642-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-021-17642-9